





Клиническая фармакология. Неотложная помощь

П Р Е Д М Е Т И С О Д Е Р Ж А Н И Е К Л И Н И Ч Е С К О Й Ф А Р М А К О Л О Г И И
Клиническое изучение лекарственных средств складывается из ряда направлений, часть из которых перекрещивается с экспериментальной фармакологией (например, изучение механизмов действия или токсичности), а часть (оценка клинической эффективности, фармакокинетика и др.) требует исключительно клинических условий. Эти направления соответствуют принятым рубрикам описания лекарственных препаратов и являются содержанием основных разделов клинической фармакологии, к которым можно отнести:
- фармакодинамику,
- фармакокинетику,
- побочное действие лекарств,
- взаимодействие лекарств,
- фармакогенетику,
- фармакоэкономику
и др.
Особый раздел, являющийся также предметом клинической фармакологии, составляют медико-юридические и деонтологические аспекты разработки, испытаний и регистрации новых лекарственных средств.
Ф А Р М А К О Д И Н А М И К А
Термином «фармакодинамика» описывается спектр фармакологических эффектов лекарственного средства («вторичная фармакологическая реакция») и механизмы действия, через которые эти эффекты опосредуются («первичная фармакологическая реакция»), то есть изменения в определенных функциях организма, возникающие под влиянием фармакологически активного вещества. В общем виде механизмы действия лекарств можно свести к их влиянию на специфические и неспецифические тканевые, клеточные или субклеточные мишени.
Вместо достаточно нейтрального термина «мишень» иногда используется понятие «специфические и неспецифические рецепторы», что вносит определенную путаницу, поскольку «рецептор» специфичен по определению.
Среди множества этих механизмов можно выделить несколько основных групп.
Группа I. Наиболее распространенным механизмом действия лекарств, применяющихся в настоящее время, вероятно, является взаимодействие с рецепторами.
Под «рецепторамами» (абортированная форма термина «клеточные рецепторы») понимаются специальные макромолекулярные клеточные структуры, функцией которых является восприятие специфических стимулов с последующим вторичным изменением активности ферментов и целенаправленным влиянием на метаболизм и функционирование клетки. В роли таких стимулов выступают адекватные для каждой группы рецепторов химические соединения - лиганды, посредством которых осуществляется передача информации от одной клетки к другой.
Специфичность лигандов для рецепторов служит тем признаком, на основе которого проводится классификация рецепторов (ГАМК-эргические, дофаминовые, и т.п.). Рецепторы чаще располагаются на поверхности клеточной мембраны (опиатные, ангиотензиновые, холинергические, адренергические, гистаминовые и др.), но могут находиться и внутри клетки, например, рецепторы к глюкокортикоидам.. Введением в организм веществ, способных вместо эндогенных лигандов связываться с рецепторами, можно направленно изменять функциональное состояние клетки, то есть управлять им.
Вещества, возбуждающие рецепторы, называются агонистами, «миметиками» или стимуляторами (агонист опиатных рецепторов - морфин, холиномиметик - карбахолин, альфа-, бета-адреностимулятор - адреналин).
Вещества, препятствующие возбуждающему действию эндогенных и экзогенных агонистов, называются антагонистами, или блокаторами: антагонист опиатных рецепторов - налоксон, блокатор м-холинорецепторов - атропин, бета-адреноблокатор - атенолол.
Применявшийся ранее термин «литик» сегодня используется для описания результирующего эффекта, заключающегося в ослаблении деятельности какой-либо системы (симпатолитический эффект) или в устранении какого-либо симптома (анксиолитическое действие).
Если же вещество, защищая рецептор от влияния агониста, само обладает стимулирующим действием, которое меньше максимально возможного, то оно относится к частичным агонистам или агонистам-антагонистам (агонист-антагонист опиатных рецепторов - налорфин, бета-адреноблоеатор с внутренней симпатомиметической активностью – окспренолол [тразикор]).
Среди различных типов рецепторов (альфа- и бета-адренэргических, гистаминовых, ГАМК-эргических, и др.) можно выделить подтипы (альфа1- и альфа2- или бета1- и бета2-адренорецепторы, H1 и H2 гистаминовые рецепторы, ГАМК A и ГАМК B рецепторы, m-, d- и k- подтипы опиатных рецепторов и т. д.), имеющие различные, иногда противоположные, эффекты и способные избирательно (селективно) реагировать на некоторые химические вещества.
Такие вещества считаются селективными (бета1-адренергическими, H2 гистаминовыми и т.п.) агонистами или антагонистами. Селективность является важной характеристикой лекарственного средства, однако в клинических условиях она в определенной мере носит условный характер. Являясь по сути не «избирательностью», а «преимущественностью», она наблюдается лишь при относительно невысоких концентрациях лекарственного вещества и исчезает при увеличении концентрации. Так, например, атенолол, имеющий более выраженный аффинитет к бета1-адренорецепторам сердца, считается кардиоселективным адреноблокатором. Тем не менее, в дозах, вызывающих клинически значимую депрессию синусового узла, он способен влиять и на бета-адренорецепторы бронхов, вызывая или усугубляя бронхоспазм.
В клинических условиях имеет значение не только относительность селективности лекарственных веществ по отношению к подтипам рецепторов, но и относительность представлений об органоспецифичности подтипов рецепторов: принято считать, что бета1-адренорецепторы имеют кардиальную органоспецифичность, а бета2-рецепторы – бронхиальную. Тем не менее, в миокарде, в частности в синусовом узле, активную функцию выполняют бета2-рецепторы, а в бронхах обнаружены и бета1-адренорецепторы.
Аналогичное замечание правомочно и в отношении селективности лекарств с другими механизмами действия – см. ниже (например, к селективно действующим на определенные изоферменты или подтипы кальциевых каналов).
На примере рецепторов можно разобрать еще два понятия, которые, как и селективность, распространяются на другие механизмы.
Первое из них – «аффинитет», являющееся количественной характеристикой степени сродства фармакологического вещества к рецептору (мишени) и определяющее скорость образования и прочность связи между ними. Высокий аффинитет позволяет лекарственному веществу уже при небольших концентрациях связываться с рецепторами (мишенями), при низком же аффинитете для достижения эффекта требуются более значительные концентрации.
Второе – «обратимость/необратимость» связи. При обратимости связывания образование и стойкость комплекса вещество-рецептор/мишень зависят от концентрации этого вещества. Ее падение приводит к освобождению рецептора (мишени), и прекращению эффекта. Иными словами, обратимая связь образуется по закону действующих масс, по которому в этом случае, например, антагонист и агонист могут вытеснять друг друга из этой связи (конкурентный антагонизм). Если же образующаяся связь с рецептором (мишенью) необратима, то восстановление прежней реакции возможно только после синтеза новых рецепторов (мишеней).
Группа II. Вторым по распространенности среди механизмов действия современных лекарственных средств можно назвать влияние на активность ферментов.
Изменяя активность ферментов, лекарственные препараты регулируют метаболизм, а соответственно, и функцию. Можно выделить ферменты, связанные с мембранами, локализующиеся в цитоплазме или субклеточных структурах, ассоциированные с синапсами или циркулирующие в крови. При этом используется почти исключительно ингибирующее действие на ферменты, например:
- антихолинэстеразные средства (неостигмин [прозерин]),
- ингибиторы МАО - неселективные (группа антидепрессантов, широко применявшаяся в клинике до недавнего времени), на смену которым пришли селективные ингибиторы изофермента МАО-А (моклобемид [аурорикс]),
- селективные ингибиторы изоферментов фосфодиэстеразы: V-го типа (силденафил [виагра]) или III-го типа (бипиридин [амринон]),
- ингибиторы мембранной АТФ-азы (сердечные гликозиды),
- неселективные ингибиторы циклооксигеназы [ЦОГ] (почти вся группа нестероидных противовоспалительных препаратов),
- селективные ингибиторы ЦОГ-2 (нестероидный противовоспалительный препарат - мелоксикам [мовалис]),
- ингибиторы протеаз (апротинин [трасилол, контрикал, гордокс]),
- ингибиторы ангиотензинпревращающего фермента а) преимущественно циркулирующего (каптоприл [капотен, ренитек, энап]) или б) как циркулирующего, так и тканевого (хинаприл [аккупро])
и многие, многие другие.
Ингибиция фермента, как и блокада рецептора (см. выше), может быть обратимой и необратимой. В последнем случае восстановление измененной лекарством функции возможно только путем синтеза новых молекул фермента, что наблюдается, например, при необратимой ингибиции ЦОГ ацетилсалициловой кислотой.
Так, необратимая ингибиция ЦОГ в клетках эндотелия сосудов не имеет клинической значимости, поскольку здесь фермент легко ресинтезируется и нарушенная продукция простациклина (обеспечивающего вазодилатацию) быстро восстанавливается. В тромбоцитах же, где отсутствует ядро, а потому невозможны синтез белка и, соответственно, ресинтез ферментов, продукция тромбаксана и агрегация тромбоцитов подавляются на все время их жизни, т. е. до 10-14 дней.
Группа III. Следующий механизм действия современных лекарственных средств заключается во взаимодействии с водными, липидными или протеиновыми компонентами клеточных мембран, и во влиянии на селективное движение ионов через них.
Благодаря уменьшению проницаемости клеточной мембраны для ионов натрия (мембраностабилизирующий эффект), действуют средства для наркоза, анестетики (прокаин [новокаин], лидокаин), противосудорожные препараты (карбамазепин [финлепсин, дифенин]), антиаритмики I, в частности Iа класса (хинидин; прокаинамид [новокаинамид], пропафенон [ритманорм]). От последних антиаритмики Iб класса отличаются не только тем, что они влияют не на активированные, а на неактивированные натриевые каналы, но и тем, что способны увеличивать проницаемость клеточной мембраны для калия.
Клинические эффекты блокаторов кальциевых каналов (верапамил [изоптин, финоптин]; нифедипин, [коринфар] и др.) обусловлены нарушением трансмембранного тока кальция. Согласно одной из гипотез стабилизация мембран тучных клеток, вызываемая, например, кромогликатом натрия [инталом], также является результатом блокады вхождения в клетку кальция.
Имеется возможность и неселективного влияния на проницаемость мембран, что опосредуется через другие механизмы (например, один из механизмов мембраностабилизирующего действия глюкокортикоидов связан с подавлением высвобождения ферментов из лизосом).
Группа IV. Достаточно распространенным механизмом действия является прямое химическое или физико-химическое взаимодействие, в том числе и включение в крупные молекулы..
Через прямое взаимодействие осуществляется первичная фармакологическая реакция большинства антидотов, которые, связываясь с субстратом, лишают его токсических свойств (например, ЭДТА - с тяжелыми металлами) или ускоряют его выведение (поливидон [гемодез]). Многие антидоты также предоставляют активные группировки для восстановления утраченных эндогенными веществами функций (так, унитиол является донатором сульфгидрильных групп, за счет чего он восстанавливает активность тиоловых ферментов, утраченную, например, при гликозидной интоксикации).
Прямое взаимодействие с соляной кислотой желудочного сока приводит к ее нейтрализации при применении системных (растворимых и всасывающихся - бикарбонат натрия) и несистемных антацидов (нерастворимых и невсасывающихся - альгельдрат c гидроокисьи магния [альмагель]). Путем прямого взаимодействия с другими веществами оказывают эффект, так называемые, вяжущие средства (танин, препараты цинка), вызывающие местное уплотнение коллоидов.
Взаимодействуя с другими химическими соединениями, некоторые лекарства могут включаться в крупные молекулы, влияя тем самым на метаболические процессы в клетке. Так, например, витамины включаются в ферменты в качестве простетических групп, а нестероидные анаболики - оротовая кислота, метилурацил, инозин [рибоксин ]- участвуют в синтезе пиримидиновых и пуриновых оснований, то есть регулируют нуклеиновый и энергетический обмены. Противоопухолевое средство мз группы антиметаболитов - флуороурацил [фторурацил] также включается в синтез нуклеиновых кислот, но, занимая место урацила, ведет к нарушению нуклеинового обмена.
У ряда препаратов клинический эффект связан с внеклеточными влияниями и достигается за счет осмотического действия – физико-химического взаимодействия с молекулами воды (осмотические мочегонные, декстран [полиглюкин, реополиглюкин], солевые слабительные). Меняя, благодаря осмотическому действию, соотношение форменных элементов и жидкой части крови, эти препараты существенно влияют на ее реологические свойства. Физико-химическое взаимодействие - механизм действия активированного угля и других сорбентов.
Группа V. Антибактериальные, противовирусные, противопаразитарные препараты нарушают различными путями метаболизм в клетках исключительно или преимущественно возбудителей различных заболеваний, минимально влияя на организм человека.
В зависимости от локализации первичной фармакодинамической реакции лекарств можно выделить:
Мстное действиеЭффекты развиваются на месте применения препарата (например, ингаляторные формы бронхолитических препаратов, антацидные средства, мази и растирки) или как побочные реакции в виде местно-раздражающего действия при, допустим, внутримышечном введении |
Резорбтивное действие Препарат доставляется в отдаленные от места введения зоны и там оказывает свои эффекты (во внутренней медицине используется наиболее часто) |
Рефлекторное действие Эффект развивается в отдаленном от зоны приложения регионе за счет местного влияния на нервные окончания без доставки самого лекарства в регионы проявления эффекта (отхаркивающие средства растительного происхождения) |
Истинное местное действие наблюдается крайне редко, поскольку в связи с частичным всасыванием может развиваться резорбтивный эффект (часто побочный, например, у ингаляционных бета2-агонистов) или присутствовать сопутствующее рефлекторное действие («разогревающие» мази).
Механизм действия, то есть первичная фармакологическая реакция, определяет вторичную фармакологическую реакцию, то есть фармакологические эффекты. В зависимости от влияния на механизмы патологических процессов фармакологические эффекты лекарственных средств могут быть классифицированы как:
- этиотропные,
- патогенетические,
- симптоматические.
При этом в первых двух случаях дополнительно возможно выделить профилактическую и заместительную терапию. Любой вид эффектов может использоваться при проведении плановой терапии и оказании ургентной помощи, хотя в последнем случае возможности этиотропной терапии ограничены. Более подробно – см. урок «Общие вопросы клинической фармакологии».
Ф А Р М А К О К И Н Е Т И К А
Выраженность первичного фармакодинамического эффекта находится в прямой зависимости от количества лекарственного вещества, достигшего мишени и связавшегося с ней. Последнее определяется динамикой его концентрации в различных средах организма. Так, зависимость между концентрацией лекарства в крови и выраженностью фармакологических эффектов в диапазоне от 20 до 80% максимального эффекта обычно приближается к прямопропорциональной. Это позволяет использовать показатели концентрации для прогнозирования эффекта, а также контроля эффективности и безопасности лечения.
Фармакокинетика изучает изменения концентрации лекарственных веществ в средах организма здорового и больного человека, а также механизмы, посредством которых осуществляются эти изменения. Если механизм действия лекарства универсален у различных биологических объектов, то фармакокинетика в связи с существенными видовыми различиями в ферментативной активности видоспецифична. Поэтому клиническое значение имеют лишь те показатели фармакокинетики лекарственных средств, которые получены не в эксперименте на животных, а непосредственно у человека. А поскольку при различных физиологических (старение, беременность и т.п.) и патологических (ожирение, лихорадка, сердечная, почечная или печеночная недостаточность и др.) состояниях наблюдаются ее существенные изменения, фармакокинетика каждого лекарства изучается как у здорового, так и у больного человека. Фармакокинетические характеристики препарата и их особенности у данного конкретного больного являются основой для выбора оптимального режима дозирования, обеспечивающего наибольшие эффективность и безопасность лечения.
Фармакокинетический цикл состоит из ряда фаз, рассматриваемых последовательно, хотя в реальном масштабе времени они протекают параллельно, лишь преобладая в те или иные моменты. Выделяют:
- поступление лекарства в организм,
- его распределение в различных средах,
- связывание с белками крови (тесно примыкающее к распределению и часто рассматриваемое вместе с ним),
- элиминация, складывающаяся из биотрансформации и выведения.
Поступление лекарства в организм.
|
При большинстве путей введения, обеспечивающих резорбтивное действие препарата, фармакологическое вещество проникает во внутреннюю среду организма путем всасывания (абсорбции), под которым понимается прохождение лекарственного вещества через биологические барьеры в кровеносную (лимфатическую) систему. Из всех энтеральных и парентеральных путей механизм всасывания отсутствует только при внутрисосудистых. |
Возможные путиЭнтеральные (т. е. через пищеварительный тракт): - оральный или внутрь, - сублингвальный, - ректальный - в любой отдел кишечной трубки с помощью зондов или фистул. Парентеральные (т. е. минуя пищеварительный тракт): - внутрисосудистые, - внутримышечный, - подкожный, - аппликационный, - ингаляционный. |
В порядке значимости различают следующие механизмы абсорбции:
- пассивная (простая) диффузия – прохождение липофильных низкомолекулярных соединений через биологические мембраны; осуществляется по градиенту концентрации и зависит от степени липофильности вещества;
- фильтрация (конвекционный транспорт) – прохождение молекул лекарственного вещества через поры мембран, что имеет достаточно ограниченное значение в связи с незначительной величиной пор (в среднем до 1 нм); кроме величины молекул фильтрация зависит от их гидрофильности, способности к диссоциации, соотношения заряда частиц и пор, а также от гидростатического, осмотического и онкотического давлений; таким путем всасываются вода, некоторые ионы и мелкие гидрофильные молекулы;
- активный транспорт – осуществляется с помощью транспортных систем клеточных мембран (специальными носителями с потреблением энергии) и может протекать против градиента концентрации; для данного механизма характерны избирательность, конкуренция двух веществ за один носитель и «насыщаемость», то есть достижение максимальной скорости процесса, лимитируемой количеством носителя и не увеличивающейся при дальнейшем повышении концентрации абсорбируемого вещества; таким способом всасываются гидрофильные полярные молекулы, ряд неорганических ионов, сахаров, аминокислот и др.
- облегченный транспорт – подобен активному транспорту, но не сопровождается потреблением энергии и может осуществляться против градиента концентрации; классическим примером этого механизма служит всасывание в кишечнике витамина В12.
- пиноцитоз – сходен с фагоцитозом; в результате инвагинации клеточной мембраны образуются вакуоли, содержащие крупные молекулы вещества; вакуоли мигрируют по цитоплазме к противоположной стенке и опорожняются наружу.
Приведенные универсальные механизмы используются не только при всасывании, но и при распределнии и выведении.
Процесс абсорбции зависит от двух групп факторов – характеризующих лекарственное вещество и определяющих состояние определенных физиологических механизмов.
Факторы, определяющие абсорбцию лекарственных веществ |
|
А. Относящиеся к веществуразмер молекулы липо/гидрофильность наличие/отсутствие электрического заряда зависимость заряда от рН среды создаваемый веществом уровень рН |
Б. Относящиеся к организмуплощадь всасывающей поверхности рН среды степень гидратации и гемоконцентрации состояние микроциркуляции |
Для практики очень важно, что изменения в физиологических функциях существенно влияют на процесс абсорбции. Так, активность самого удобного и широко применяемого пути введения – внутрь («per os») подвержена значительным колебаниям в связи с возрастом (и возрастными изменениями рН), временем приема пищи, состоянием моторики и скоростью эвакуации из желудка и т. п. Особо необходимо подчеркнуть, что все пути введения, подразумевающие использование механизмов всасывания, лимитируют или исключают этот процесс при нарушениях гидратации/гемоконцентрации и микроциркуляции. А поскольку степень микроциркуляторных расстройств напрямую зависит от стрессорности ситуации, при многих ургентных состояниях методом выбора оказывается внутривенный путь введения (но не внутримышечный или подкожный, которые требуют всасывания и зависят от состояния микроциркуляции).
Нужно отметить, что в реальных условиях оказания неотложной помощи инъекционный путь введения используется излишне активно в связи с убежденностью, что он обеспечивает наибольшую скорость наступления эффекта. Это не совсем так. Многие препараты, применяемые сублингвально, позволяют добиться необходимого эффекта в адекватные сроки – быстродействующие нитраты, нифедипин [коринфар], каптоприл [капотен], прорпанолол [обзидан, анаприлин], клонидин [клофелин, гемитон] и др. Более того, в случае системного применения глюкокортикоидов у больных с тяжелыми и угрожающими жизни приступами бронхиальной астмы назначение преднизолона внутрь более эффективно, чем внутривенно. С другой стороны отказ от применения ряда лекарств внутрь обусловлен не соображениями времени получения эффекта, а особенностями этих средств – либо их низкой всасываемостью (строфантин, стрептомицин), либо их изменениями под влиянием факторов ЖКТ – неоптимальный уровень рН, агрессивные для вещества ферменты и т. п. (инсулин, окситоцин, многие антибактериальные средства). Кроме этого, внутрь не назначаются вещества, оказывающие сильное раздражающее действие (противораковые средства).
При приеме внутрь имеется еще один фактор, ограничивающий использование ряда препаратов – печеночный метаболизм. Лекарственное вещество, поступая из ЖКТ по воротной вене в печень, подвергается ферментативному разрушению, в связи с чем в системный кровоток попадает лишь часть (иногда незначительная часть) принятой дозы. Данный феномен носит название «эффект первого прохождения через печень» или «пресистемная элиминация».
Пресистемная элиминация действительно в большинстве случаев обусловлена эффектом первого прохождения. Однако она не ограничивается последним и складывается из всех «потерь» лекарственного вещества с момента поступления в ротовую полость и до попадания в системный кровоток. Например, разрушение инсулина или тиамина происходит в кишечнике под действием соответствующих ферментов.
Этот эффект характерен для быстро метаболизирующихся средств и при значительной выраженности исключает возможность приема соответствующего препарата внутрь (например, лидокаин). В других случаях его можно корригировать увеличением дозы, которая оказывается значительно выше, чем при внутривенном введении (верапамил [изиптин, финоптин], морфин, пропранолол [анаприлин, обзидан]). Весьма демонстративным в этом плане является пример нитратов. Эффект первого прохождения через печень у нитроглицерина [нитроглицерин, нитрогранулонг, нитронг, сустак и т. д.] значительно превышает 80% дозы (по некоторым данным достигает 95-97%), что объясняет их наивысшую эффективность при назначении «в обход» печени (сублингвально или минимальные дозы внутривенно). Следующее поколение нитратов представлено изосорбидом динитратом [нитросорбид, кардикет и т. п.], эффект первого прохождения у которого не достигает 80%, что обеспечивает их более высокую эффективность. Наиболее же перспективно использование современного нитрата – изосорбида мононитрата, подвергающегося минимальной пресистемной элиминации.
Таким образом, при внесосудистом введении лекарств только часть дозы достигает системного кровотока (вследствие неполного всасывания, разрушения в месте введения под влиянием специфических и неспецифических ферментов, неоптимальной рН, а при внутреннем применении и в результате эффекта первого прохождения). Этот феномен описывается термином «системная биодоступность» или просто «биодоступность» (устар. – «биоусвояемость»). Системная биодоступность измеряется в процентах и отражает ту часть дозы, которая достигла системного кровотока.
Биодоступность может колебаться в широких пределах при различных изменениях функций. Например, она падает практически до нуля при шоке или возрастает при портальной гипертензии с развитием порто-кавальных анастамозах, позволяющим веществам с высоким эффектом первого прохождения достигать системного кровотока при приеме внутрь, минуя печень. Она может меняться при варьировании дозы. Кроме особенностей больного и характеристик лекарственного вещества, биодоступность определяется лекарственной формой (раствор или суспензия; таблетка, таблетка в оболочке, капли или сироп и т. д.) или конкретным лекарственным препаратом, выпускаемым различными фирмами. Сопоставление биодоступности вещества из различных лекарственных форм или коммерческих препаратов позволяет оценить «биэквивалентность» этих форм или препаратов. То есть по сути биоэквивалентность представляет собой сравнительную биодоступность.
Необходимо подчеркнуть, что при использовании в лечебных целях резорбтивного эффекта низкая биодоступность является отрицательной характеристикой препарата. В случаях же, когда с системной биодоступностью связаны побочные эффекты (например у ингаляционных бета2-агонистов), предпринимаются специальные меры для ее уменьшения (дозированные ингаляторы используются со спейсерами или применяется наиболее безопасная сегодня терапия – небулайзерная).
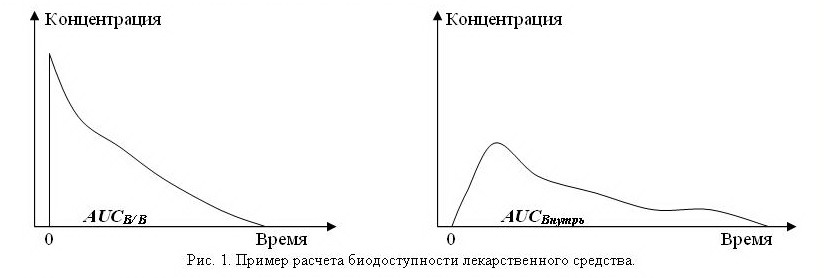
Определение биодоступности осуществляется с помощью сопоставления AUC (Area Under Curve – площадь под кривой изменения плазменной концентрации во времени), определяемой при введении одной и той же дозы внутривенно и другим путем (рис. 1). При этом биодоступность при внутривенном введении принимается за 100%.

Рассчитав AUC для внутривенного и другого пути введения одной и той же дозы, можно определить биодоступность по формуле:
СИСТЕМНАЯ БИОДОСТУПНОСТЬ = 100(AUCВнутрь/ AUCВ/В)%
При приеме препаратов внутрь имеется еще один важнейший фактор, влияющий на скорость абсорбции и биодоступность в целом – взаимодействие с пищей. Причем оказывается, что под влиянием пищи эти две характеристики могут изменяться независимо друг от друга, а иногда и разнонаправлено. Так, если у амоксициллина, ацетилсалициловой кислоты прием пищи замедляет всасывание и уменьшает биодоступность, то при замедлении всасывания эритромицина пища может даже увеличить его биодоступность. Правильное соотношение приемов лекарства и пищи может не только повысить эффективность лечения, но и предупредить развитие побочных реакций.
Скорость поступления препарата в системный кровоток служит одним из решающих факторов, определяющих величину максимальной плазменной концентрации лекарственного вещества (Сmax) и время достижения этой концентрации (Tmax)
СВЯЗЫВАНИЕ С БЕЛКАМИ КРОВИ
Многие лекарственные вещества обладают выраженным физико-химическим сродством к макромолекулам, в связи с чем, попав в кровь или лимфу, они связываются с белками. Это имеет существенное значение, поскольку только свободная фракция вещества фармакологически активна. Она способна проникать через клеточные мембраны, влиять на специфические мишени (см. выше), подвергаться превращениям под влиянием ферментов или экскретироваться из организма. Свободная и связанная фракции находятся в равновесии: в связанном виде вещество циркулирует в крови до тех пор, пока концентрация свободной фраккции не снизится вследствие тех или иных процессов, после чего роисходит высвобождение его части, что обеспечивает стабильность плазменной концентрации. Иными словами, связавшись с белками крови, препарат образует депо.
Среди белков крови наибольшую роль в образовании комплексов с лекарствами играет альбумин (который связывает салицилаты, пенициллины, сульфониламиды и многие др., главным образом, слабые кислоты). Однако в этом процессе участвуют и другие компоненты крови – глобулины, липопротеиды, a1-кислый гликопротеид и даже форменные элементы. При этом некоторые вещества связываются одновременно с несколькими структурами. Например, хлорпромазин [аминазин] связывается с липопротеидами и эритроцитами, трициклический антидепрессант имипрамин [мелипрамин] и хинидин – с липопротеидами, a1-кислым гликопротеидом и эритроцитами.
Связывание с белками приобретает клиническое значение тогда, когда оно превышает 80-90%. Так, уменьшение связи с белками с 98% до 96% способно увеличить свободную фракцию на 100%, что чревато развитием передозировки. Такая ситуация может развиться при различных физиологических и патологических состояниях, при которых уменьшается количество белка в крови (например, новорожденные и особенно недоношенные, пожилые люди, истощенные больные, пациенты с нарушенной белковосинтетической функцией). Так, для значительного увеличения свободной фракции фенитоина [дифенина] достаточно снижения сывороточного альбумина до 30 г/л (нижняя граница нормы – 33 г/л); этот эффект у фуросемида [лазикса] становится значимым при снижении альбумина до 20 г/л. Одной из причин различных реакций на изменения концентрации белка у разных препаратов служат особенности их кажущегося объема распределения (см. ниже).
РАСПРЕДЕЛЕНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ В ОРГАНИЗМЕ
Большинство лекарств, попав в системный кровоток, разносятся по всему организму, проникая в интерстициальные пространства различных органов и далее – в клетки тканей. Процесс распределения продолжается до тех пор, пока скорость движения лекарственного вещества в ткани не сравняется со скоростью его возвращения из ткани в кровоток. При равенстве этих скоростей возникает состояние, считаемое устойчивым (steady state), а концентрация вещества в крови в это время называется равновесной (CSS).
Распределение препаратов в организме никогда не бывает равномерным, что зависит от ряда физиологических (патофизиологических) и фармакологических факторов.
К первым относятся:
- интенсивность регионарного кровотока в физиологических условиях (наиболее активно кровоснабжаются такие органы как сердце, печень, почки; скорость кровоснабжения желез внутренней секреции почти в 100 раз больше, чем эквивалентного количества жировой ткани);
- проницаемость мембран и соответствующих барьеров (например, гематоэнцефалического, плацентарного) для данного вещества в норме и при патологии (так, в нормальных условиях пенициллин не проникает через гематоэнцефалический барьер, который при менингите становится для него проницаемым);
- нарушения гемодинамики и микроциркуляции при стрессе, шоке, хронической сердечной недостаточности, в результате чего уменьшается кровенаполнение интенсивно снабжаемых кровью органов (тормозятся инактивация препарата в печени, экскреция с мочой);
- наличие в полостях застойных и воспалительных выпотов, в которых способны накапливаться гидрофильные лекарственные вещества).
Среди свойств лекарственного вещества, определяющих характер распределения можно выделить:
- факторы, от которых зависит способность вещества к абсорбции (преодаление биологических барьеров в процессе распределения происходит по тем же законам, что и при всасывании) – см. выше;
- сродство вещества к определенным тканям, что обеспечивает преимущественное накопления лекарства в них.
Необходимо оговориться. Наивысшие концентрации лекарства далеко не всегда наблюдаются в органах, где проявляется основной фармакологический эффект, поскольку, накопившись в ткани или органе, вещество может:
связаться со специфическими мишенями, что и определяет эффект;
связаться с неспецифическими мишенями (обычно с белками тканей);
остаться в свободной растворенной форме;
подвергнуться биотрансформации;
реабсорбироваться в кровоток в неизмененном виде;
экскретироваться в неизмененном виде (например, с грудным молоком).
В связи с неравномерностью распределения и возможностью создания в отдельных регионах очень высоких концентраций при низких в других предложен интегральный показатель, характеризующий распределение лекарства с учетом всех факторов, влияющих на этот процесс. Таким показателем служит объем распределения или кажущийся (условный) объем распределения под которым понимается гипотетический объем жидкости, в котором введенная доза создаст концентрацию, равную концентрации в крови. Иными словами, весь организм представляется одной камерой. Причем малый объем распределения свидетельствует о том, что распределение препарата минимально и он преимущественно остается в сосудистом русле. И наоборот, большой объем распределения характеризует активное накопление препарата во всех органах, что создает высокие тканевые концентрации и приводит к тому, что кажущийся объем распределения превышает не только объем жидкости, имеющейся в организме (примерно 42 л у взрослого человека массой 70 кг), но и объем всего тела. Объем распределения выражается в литрах на единицу массы тела (относительный объем распределения), нпример, он составляет:
у метиленового синего – 0,043 л/кг,
у фуросемида [лазикса] – 0,11 л/кг,
у дигоксина – 8,6 л/кг,
у хлорохина - 171,4 л/кг.
Из приведенных примеров видно, что при массе 70 кг необходимый для распределения метиленового синего объем ограничивается 3 л (0,043 л/кг ´ 70 кг), то есть объемом плазмы. Для дигоксина же необходим объем в 600 л, что почти в 9 раз превышает объем тела массой 70 кг, т. е. тканевые концентрации во много раз превышают плазменную.
Клиническое значение имеет соотношение объема распределения связывания препарата с циркулирующими белками. При клинически значимом связывании препарата с белком гипопротеинемия чревата существенным увеличением свободной фрактции лекарственного вещества в крови в случае небольшого объема распределения (например, у фуросемида [лазикса], у которого связь с белком составляет 91- 99%, а объем распределения при массе 70 кг – 7,7 л).Если же при тех же условиях объем распределения велик, т. е. основная часть препарата связана в тканях, то его концентрация в крови не должна меняться существенно (например, у пропранолола [анаприлина, обзидана] или верапамила [изоптина, финоптина], у которых связь с белком составляет, соответственно, 90-95 и 90%, а объем распределения 280 и 315 л).
Показатель кажущегося объема распределения служит одним из критериев дозирования препарата в той или иной ситуации. Например, изменение с возрастом относительного объема жидкости в организме часто требует использования у детей более высоких относительных доз (на килограмм массы тела), чем у взрослых, и меньших относительных доз у лиц пожилого и старческого возраста.
Важно подчеркнуть, что изменения фракции, связанной с белком, в патологических условиях могут протекать в обе стороны. Например, у хинидина этот показатель, составляющий в норме 87-92%, при застойной сердечной недостаточности снижается до 82%, а при хронической дыхательной недостаточности возрастает до 96%. При инфаркте миокарда происходит повышение a1-кислого гликопротеина, что способствует усиленному связыванию верапамила [изоптина, финоптина], дизопирамида [ритмодана], лидокаина, хинидина и др. и в результате с 5 по 12 день болезни отношение связанного и свободного дизопирамида возрастает в 3 раза.
Несмотря на неравномерность распределения, фармакологический эффект препарата, как уже указывалось выше, существенно зависит от плазменной концентрации лекарственного вещества. Для каждого средства можно выделить уровни концентрации, которые будут соответствовать определенному действию:
- концентрация, при которой лечебный эффект развивается у половины испытуемых, принимается за минимальный Терапевтический Уровень (МТУ);
- концентрация, при которой отмечаются первые проявления токсического побочного действия, называется Минимальной Токсической Концентрацией (МТК);
- концентрации между МТУ и МТК составляют Терапевтический Диапазон;
- середина терапевтического диапазона считается Средней Терапевтической Концентрацией, к созданию и поддержанию которой стремятся при лекарственной терапии;
- отношение верхней границы терапевтического диапазона к нижней (МТК/МТУ) служит показателем безопасности препарата и называется Терапевтической ШиротоЙ.
Контроль плазменной концентрации позволяет обеспечить наибольшую эффективность и безопасность терапии, особенно у препаратов с узким терапевтическим диапазоном и малой терапевтической широтой. Однако, чем выше аффинитет, тем меньшую роль играет концентрация, и большую роль приобретает общая доза. Для иллюстрации этого положения можно привести два примера.
- В целях достижения наибольшей простоты предположим, что аминофиллин [эуфиллин] обладает только двумя эффектами, независимыми друг от друга: бронхолитическим и «кардиотропным» (под последним подразумевается способность препарата повышать потребление кислорода миокардом и, главное, аритмогенное действие). В связи с угрозой неблагоприятного влияния на функции миокарда этот препарат требует особой осторожности при лечении пожилых больных и пациентов с заболеваниями сердца (ИБС). Между тем, вероятность развития лечебного и побочного эффектов неодикова и для последнего она может быть еще уменьшена, если учесть фармакокинетические особенности препарата. Дело в том, что аффинитет аминофиллина [эуфиллина] к миокарду невелик, и для развития кардиотропных эффектов необходимы большие его концентрации. С другой стороны, аффинитет данного средства к ткани бронхов столь значителен, что даже при невысоких его концентрациях в крови достаточное его количество соединится с мишенью. Следовательно, если препарат вводить внутривенно медленно, то низкая концентрация не позволит ему фиксироваться на миокарде, но не помешает бронхолитическому эффекту. Таким образом, рекомендуемое везде медленное (10 мл 2,4% раствора за 3-5 мин или, лучше, капельное) введение аминофиллина [эуфиллина] обеспечивает максимальную безопасность при сохранении эффективности.
- Столь же схематично рассмотрим действие фуросемида [лазикса]. Его диуретический эффект обесловлен влиянием на петлю Генле в почке, аффинитет к которой у него относительно невысок. Что же касается гипотензивного эффекта, то, несмотря на привычное объяснение его возникновения снижением волемии, главную роль в его развитии играет непосредственное вазодилатирующее влияние. При этом аффинитет диуретика фуросемида [лазикса] к сосудистой стенке (в первую очередь венозной и легочной артерии) весьма значителен. В связи с этим на фоне медленного внутривенного введения фуросемида [лазикса] развивается преимущественно гипотензивный эффект (пропорциональный дозе) при минимальном диуретическом действии. Для получения же мочегонного действия препарат необходимо вводить внутривенно быстро. Что же касается нередких рекомендаций медленного его введения для предупреждения коллапса, то, поскольку гипотензивный эффект зависит от дозы, скорость введения в его развитии роли не играет.
ЭЛИМИНАЦИЯ
По характеру элиминации из организма все лекарства могут быть разделены на две группы. Часть (в основном высокогидрофильные ионизированные соединения) лекарственных веществ, и таких меньшинство, выделяются в неизмененном виде. Другие – подвергаются химическим превращениям, в результате которых в конечном итоге образуются более полярные молекулы, которые быстро выводятся с мочой и желчью. В отличие от веществ, свойственных организму, химические преобразования ксенобиотиков (чужеродных для организма веществ), к коим относятся и лекарства, принято называть биотрансформацией.
А. Биотрансформация (метаболизм)
Основной биологической идеей биотрансформации являятся освобождение организма от ксенобиотика либо путем его утилизации в качестве энергетического или пластического субстрата, либо путем его перевода в форму, удобную для выведения. Поэтому соответствующие биохимические процессы можно было бы рассматривать как систему дезинтоксикации, однако такой взгляд был бы слишком упрощенным.
- Во-первых, хорошо известно, что токсичность многих ксенобиотиков обусловлена не самим веществом, а продуктами его биотрансформации. Это в полной мере относится и к лекарствам. Так, например, токсичный ксилидид моноэтилглицина, образуется из лидокаина, что и определяет токсичность последнего.
- Во-вторых, большое число препаратов имеет активные дериваты (метаболиты), фармакологическое действие которых, сравнимо или значительно превышает таковое у первоначального вещества. Так, активность 4-гидрокси-пропранолола, образующегося в печени при первом прохождении, сравнима с активностью самого пропранолола [анаприлина, обзидана]; однако, в связи с тем, что первый обладает меньшим периодом полувыведения (см. ниже), разные способы введения данного препарата обеспечивают разную его эффективность. На группе ингибиторов АПФ, действие которых почти всегда связано с активными дериватами (из рутинно применяющихся исключение составляет каптоприл [капртен]), можно продемонстрировать увеличение активности фармакологического вещества в процессе биотрансформации. Так, эналаприл [ренитек, энап], гидрализуясь, превращается в почти на порядок более активный эналаприлат. В ряде случаев одни лекарства могут превращаться, обычно в небольшой части дозы, в вещества, используемые как другие лекарства: например, кодеин способен трансформироваться в морфин или (у новорожденных) теофиллин в кофеин.
- В-третьих, множество лекарств оказывают фармакологический эффект не сами, а через продукты биотрансформации. Например, действие нитратов опосредуется через высвобождающийся вазодилатирующий фактор – NO. Целая группа «лекарств» представляет собой как бы транспортные вещества, которые обеспечивают лишь процесс доставки, сами являясь по сути «пролекарствами», активность которых обеспечивается их дериватами. Со временем иногда появляется возможность отказаться от них, перейдя к выпуску непосредственно активного начала: вместо не применяющегося сегодня фенацетина (пролекарства) широко используется его активный дериват – парацетамол.
В порядке убывания значимости органы и ткани, принимающие участие в биотрансформации, можно расположить следующим образом: печень, желудок, кишечник, почки, легкие, кожа, мозг. Необходимо подчеркнуть, что в этом процессе могут принимать участие и другие структуры – надпочечники, гладкие и поперечнополосатые мышцы, эндотелий сосудов, кровь и т. п.
В реакциях биотрансформации можно выделить два этапа (две фазы), каждый из которых может иметь и самостоятельное значение:
|
Реакции I фазы (несинтетические)
|
Реакции II фазы (синтетические) – конъюгация с:
|
Многие лекарства одновременно подвергаются нескольким реакциям каждой из фаз. Такое дублирование обеспечивает высокую надежность деятельности системы в целом. При этом конечные продукты биотрансформации могут различаться при различном состоянии отдельных систем организма, например, у детей разного возраста.
Среди реакций I фазы (несинтетических) ключевую роль играет система изоферментов цитохрома Р450 – главная окисляющая система организма, связанная с эндоплпзматическим ретикулумом (эндоплазматическая или микросомальная система). Ее наибольшая активность отмечается в печени. Важнейшими свойствами микросомальной системы изоферментов цитохрома Р450 являются:
- возможность биотрансформировать практически все известные химические соединения;
- способность связывать молекулярный кислород;
- высокая индуктивность (повышение активности фермента под влиянием внешних факторов).
Возможна селективная индукция определенных изоферментов и более или менее неселективная индукция. Последняя может возникать под влиянием алкоголя и ингредиентов табачного дыма, в связи с чем у лиц с так называемыми вредными привычками может существенно снижаться эффективность множества препаратов. Важно подчеркнуть возможность не только индукции, но и ингибиции ферментов этой системы (например, уксусным альдегидом, образующимся при восстановлении этанола под влиянием алкогольдегидрогеназы).
Необходимо отметить, что при длительном применении ряда лекарственных препаратов (дифенгидрамин [димедрол]; фенобарбитал, включая комбинированные препараты [андипал, беллатаминал, валокордин, теофедрин и др.]; фенотиазины – хлорпромазин [аминазин] и др., прометазин [дипразин, пипольфен], триметоприм, включая комбинированные препараты – ко-тримоксазол [бактрим, бисептол, септрин и др.]) под влиянием изоферментов системы цитохрома Р450 могут образовываться так называемые эпоксиды и азотосодержащие оксиды, обладающие способностью повреждать клеточные мембраны, структурные и мембранные белки, нарушать синтез нуклеиновых кислот, вызывать канцерогенез, мута- и тератогенез.
Реакции II фазы (синтетические) завершают дезинтоксикацию, поскольку, как правило, только после конъюгации образуются неактивные или малоактивные соединения. В отличие рекций I фазы, которые протекают без затрат энергии, образование сложных молекул во II фазу обычно идет с потреблением энергии.
Особо необходимо подчеркнуть, что в случаях, когда каждая из фаз выступает в качестве самостоятельной биотрансформирующей системы (например, окисление алкоголя до углекислого газа и воды или ацетилирование сульфониламидов), высокая активность одной из них обычно сочетается с низкой активностью другой (что генетически детерминировано). Так, среди коренных северных народов очень высок процент «быстрых ацетиляторов», что сочетается с низкой способностью к окислению ксенобиотиков и плохой переносимостью алкоголя.
Понятно, что биохимические процессы, протекающие с участием ферментов (как реакции I фазы, так и II фазы), зависят от функционального состояния множества систем организма – от наличия или отсутствия патологических процессов в том или ином органе, от характера оксигенации тканей, от белковосинтетической функции в целом и активности синтеза ферментов в частности. Известно, что активность синтеза белка и ферментов меняется с возрастом. Последним объясняется снижение биотрансформации ксенобиотиков у лиц старших возрастных групп (что требует особой осторожности при дозировании лекарств у них) и существенные особенности биотрансформации лекарств в разные периоды созревания детей (с чем связаны известные ограничения применения определенных препаратов в педиатрии и детские дозы).
Наиболее активно биотрансформация протекает в печени. В связи с этим в зависимости от скорости биохимических превращений все лекарства могут быть разделены на препараты с высоким печеночным клиренсом и на средства с низким печеночным клиренсом.
Б. Выведение (экскреция)
Экскретироваться из организма лекарства могут с любыми жидкостями (моча, слюна, пот, желчь и т. д.), а летучие (газообразные средства для наркоза, эфирные масла, например, камфора) – и с выдыхаемым воздухом. Однако на практике для подавляющего большинства лекарственных средств в качестве пути выведения клиническое значение имеют почки и ЖКТ. В зависимости от гидро- или липофильности, спосбности фильтроваться, секретироваться и реабсорбироваться в почках, способности секретироваться в желчь и абсорбироваться в кишечнике лекарственные вещества и их дериваты (если средство подверглось биотрансформации) выделяются одним из двух основных путей.
Как правило, почками выделяются гидрофильные ионизированные и полярные молекулы (исходные вещества и продукты биотрансформации. А поскольку степень ионизации изменяется при различных значениях рН, почечная экскреция лекарств может существенно зависеть от рН мочи. Важно, что существуют лекарственные средства, сами влияющие на рН мочи, и это может создавать условия для их взаимодействия с другими препаратами.
Ощелачивание мочи вызывают бикарбонат натрия (при ощелачивании крови), большие объемы изотонического раствора хлорида натрия и ацетазоламид [диакарб] (в обоих случаях – при закислении крови), тиазидные мочегонные – гидрохлоротиазид [гипотиазид] (при возможности развития гипохлоремического алкалоза).
Ацидотические сдвиги в моче вызывают хлорид аммония, салицилаты, аскорбиновая кислота.
Почки представляют собой основной путь выведения лекарственных веществ, однако возможности почечной экскреции ограничены, поскольку высокомолекулярные вещества (> 60 000) практически не фильтруются, а липофильные молекулы, попав в первичную мочу, активно реабсорбируются методом простой диффузии.
Вторым по значимости путем экскреции служит ЖКТ. Весь объем выделяющегося таким образом лекарства состоит из нескольких фракций: а) часть дозы, не всосавшаяся в ЖКТ (в неизмененном виде), б) неизмененное вещество и, чаще, его дериваты, секретированные печенью в желчь и экскретированные с желчью в просвет кишки, в) часть дозы, биотрансормировавшаяся в желудке и кишечнике (в виде дериватов), г) неизмененное вещество или его дериваты, экскретированные стенкой желудка или кишки. Наибольшее клиническое значение имеют первые две фракции, поскольку именно они определяют наибольший объем экскреции лекарства этим путем. Экскрецию с желчью не ограничивют высокая молекулярная масса и связывание с белком, более того, этим путем экскретируются главным образом молекулы с массой, превышающей 300. Между тем, для большинства препаратов этот путь играет вспомогательную роль.
Особо необходимо подчеркнуть, экскретированное с желчью в просвет кишки вещество способно вновь абсорбироваться. Это касается в первую очередь конъюгатов, расщепляющихся под влиянием кишечной флоры, в результате чего высвободившееся первоначальное вещество может вновь достигать печень и вновь частично попадать в системный кровоток, а частично экскретироваться – энтерогепатическая циркуляция или печеночная рециркуляция, в связи с чем на фоне уменьшающейся плазменной концентрации могут появляться новые пики. Возможность реабсорбции экскретированного в просвет кишки вещества играет важную роль при интоксикациях и часто требует назначения энтеросорбентов вне зависимости от пути введения препарата.
Элиминацию в целом характеризует показатель, называемый клиренсом (Clобщ.), под которым понимается объем крови (плазмы, сыворотки), полностью освобождаемый от данного вещества за единицу времени. Этот показатель в практической деятельности важен для расчета поддерживающей дозы (Dподдерж.) трудно управляемых лекарств, например, дигоксина или теофиллина. Поддерживающая доза должна обеспечивать равенство между скоростью выведения и скоростью поступления препарата, то есть превратить достигнутую концентрацию в равновесную (CSS):
Dподдерж. = CSS´ Clобщ.
Для большинства лекарств общий клиренс – величина постоянная, не зависящая от концентрации. Однако у других (фенитоин [дифенин], ацетилсалициловая кислота [аспирин]) клиренс непостоянен, и элиминация – процесс назыщаемый, зависящий от дозы и концентрации. В то же время органный клиренс (почечный, печеночный) обычно зависит либо от скорости кровотока через этот орган, либо от концентрации вещества в крови, либо от обоих этих факторов. Это зависит от активности биотрансформации вещества, имеющегося запаса соответствующих ферментов, степени связывания лекарства с белками. Во всяком случае, назначая препарат, выводящийся каким-либо путем, необходимо хотя бы на качественном уровне оценить кровоток в органе, обеспечивающем этот путь – кровоснабжение почек и печени при шоке, застойной сердечной недостаточности, состояние кровообращения в печени при алкогольном циррозе печени и т. п.
Еще одна важная фармакокинетическая характеристика препарата – период полуэлиминации – Т1/2 (период полувыведения, период полужизни – менее удачные термины) – время, за которое плазменная концентрация вещества снижается в 2 раза. Он выражает связь между объемом распределения и клиренсом и зависит от обоих. На практике период полуэлиминации интересен, пожалуй, только с одной стороны. При повторном приеме препарата в одной и той же дозе через более или менее одинаковые промежутки времени, соизмеримые с периодом полуэлиминации, его концентрация в крови будет постепенно нарастать, пока скорость элиминации не достигнет скорости поступления. С этого момента концентрация превратится в равновесную. Имеется возможность заранее расчитать время наступления этого момента. При указанных условиях оно равно 5-7 периодам полуэлиминации.
П О Б О Ч Н О Е Д Е Й С Т В И Е Л Е К А Р С Т В
По определению экспертов ВОЗ побочным действием считается "любая вредная и нежелательная для организма реакция на лекарственное средство...". Практически нет такого лекарственного средства, обладающего клиническими эффектами, которое не могло бы вызвать какой-либо вредной реакции. Правда, проявляются эти побочные действия с разной частотой, и могут быть столь редки, что для их проявления необходимы многие годы, иногда десятилетий, и десятки миллионов наблюдений.
|
Считается, что побочные реакции развиваются у 4-29% лиц, принимающих лекарства, служат причиной 2-3% обращений к врачу, и до 5% госпитализаций, а в 3% случаев требуют проведения интенсивной терапии. До 12% случаев побочного действия у госпитальных больных служат причиной увеличения срока госпитализации, до 0,27% – причиной смерти (до 1,5% при внутривенном введении, преимущественно, у тяжелых больных). |
К развитию побочных эффектов предрасполагают:
|
Побочные эффекты чаще развиваются в начале лечения (на 1-10 день), хотя возможны и более поздние реакции, например, для развития гинекомастии при применении метоклопрамида [церукала] или спиронолактона [верошпирона] необходим длительный прием.
Приводимые данные о частоте побочных реакций при различных патологических состояниях и при использовании различных групп препаратов требуют дополнительного анализа, поскольку, как правило, не учитывается частота самих патологических состояний и частота применения соответствующих лекарств.
Среди причин развития побочного действия можно выделить
а) причины, связанные с лекарством и
б) причины, связанные с организмом больного.
С причинами второй группы связана идиосинкразия – повышенная чувствительность организма к определенным химическим вещества, проявляющаяся необычной реакцией на лекарство. Хотя и считается, что идиосинкразия может быть врожденной (генетически детерминированной) и приобретенной (следствие заболеваний), как правило, речь идет о генетических аномалиях, приводящих к ферментопатиям. Так, побочное действие, известное как «аспириновая желтуха» (гемолиз при применении аспирина, других салицилатов, а также противомалярийных средств, сульфаниламидов, нитрофуранов), развивается чаще всего при генетически детерминированном дефиците глюкозо-6-фосфатдегидрогенизы.
Побочные действия лекарств можно сгруппировать следующим образом:
А. Сопутствующие фармакологические эффекты
а) функционально-метаболические или фармакодинамические, связанные с фармакологическими свойствами и определяющиеся механизмом действия лекарства (бронхоспазм, вызываемый бета-блокаторами, атония желудочно-кишечного тракта и мочевыводящих путей под действием антагонистов кальция, снижение сексуальной потенции бета-блокаторами или верошпироном, дисбактериоз при применении антибактериальных средств и т.д.); особую форму этого вида побочных действий представляет "синдром обкрадывания", заключающийся в ухудшении состояния одних систем или отдельных звеньев, сопряженном с достижением положительных сдвигов в других (например, улучшение кровоснабжения отдельных зон головного мозга за счет ишемизированных участков при применении папаверина или эналаприла [ренитека, энапа]);
б) токсические (обратимые и необратимые), определяющиеся химической структурой лекарственного вещества и потому однотипные у производных одних и тех же соединений (например, у антибиотиков группы аминогликозидов); они, как правило, не имеют прямого отношения к фармакологическим эффектам и связаны с плазменной концентрацией, зависящей в первую очередь от применяемой дозы (абсолютная передозировка) или от функционирования путей инактивации и выведения (относительная передозировка), однако в ряде случаев токсические эффекты могут проявиться при терапевтических концентрациях, что обусловлено повышенной чувствительностью к препарату; токсическое действие химических веществ обычно органотропно (гепато-, миело-, нефро-, ото- и т.д.), поэтому патологический процесс в каком-либо органе может способствовать проявлению побочных реакций (например, ототоксический эффект гентамицина, применяемого в небольших дозах, у лиц с начинающимся кохлеарным невритом); токсическое действие может зависеть не только от концентрации, но и от длительности применения препарата;
в) мутагенное, онкогенное, тератогенное, эмбриотоксическое – теснопримыкают к токсическим и развиваются по подобным принципам.
Б. Аллергические (иммунологические) реакции немедленного и замедленного типов
Аллергические реакции зависят не столько от свойств лекарственного препарата, сколько от состояния иммунологической реактивности организма. Они имеют две принципиальные особенности:
а) независимость от дозы и
б) непредсказуемость.
Если для сенсибилизации организма доза играет значительную роль, также как повторяемость контакта пациента с аллергеном, длительность интервалов и др., то величина разрешающей дозы не влияет ни на вероятность развития аллергической реакции, ни на их выраженность (большинстве типов реакций). Непредсказуемость аллергичесих реакций заключается в том, что благополучное использование лекарства в прошлом не может служить гарантией безопасности его применения в настоящем. Более того, на практике нередко не удается установить наличие предшествовавшего контакта больного с данным веществом, однако при первом же назначении лекарства существует угроза развития аллергической реакции.
Иммунопатологические побочные эффекты могут иметь в своей основе реакции четырех различных типов, однако основные закономерности возникновения у них общие. Их развитию нередко способствуют некоторые дополнительные неблагоприятные факторы - погрешности в диете, употребление алкоголя и др.
В. Лекарственная зависимость
- Синдром отмены возникает, как правило, при внезапном прекращении приема лекарства. Этот синдром может проявляться тремя клиническими вариантами:
- обострением основного заболевания, ремиссия которого была достигнута на фоне терапии (например, при резкой отмене антидепрессантов, антиаритмических или антиангинальных средств, H2-блокаторов);
- развитием недостаточности системы, функцию которой замещало применявшееся средство (недостаточность коры надпочечников после отмены глюкокортикоидных гормонов, тромбозы после отмены гепарина и т.д.);
- собственно «синдромом отмены» - развитием нового патологического состояния, не наблюдавшегося ни до начала, ни в процессе приема данного лекарственного средства (симпато-адреналовые кризы после отмены бета-блокаторов, генерализованная воспалительная реакция мезенхимальной ткани вплоть до волчаночного синдром при отмене глюкокортикоидов); одним из вариантов собственно синдрома отмены является абстинентный синдром как одно из самых наглядных проявлений лекарственной зависимости.
- Пристрастие складывается из физической, психической и психологической зависимости и проявляется различными вариантами синдрома отмены при прекращении применения лекарства. Наиболее типична зависимость к опиоидам – наркомания. Однако часто развиваются различные варианты зависимости или их сочетания к лекарственным препаратам с не столь выраженными психотропными (психическая зависимость) и соматическими (физическая зависимость) эффектами. Так, хорошо известны больные бронхиальной астмой, нуждающиеся во внутривенном и при этом быстром (!) введении аминофиллина [эуфиллина], не в связи с бронхообструкцией, а для получения соответствующих физических ощущений и психических эффектов. Пристрастие может формироваться и на основе только психологической зависимости от препарата, не обладающего психофармакологической активностью. К такой форме побочного действия относится, например, вариант головной боли, связанный с постоянным приемом ненаркотических анальгетиков.
Г. Лекарственная устойчивость
К побочным действиям лекарств иногда относят также привыкание, то есть снижение чувствительности организма к препарату, требующее прогрессивного увеличения дозы, и лекарственную устойчивость, то есть отсутствие эффекта от лекарства, не преодолевающееся увеличением дозы или требующее такой дозы, при которой всегда проявляются опасные нежелательные эффекты. Однако привыкание, также как первичная (врожденная) устойчивость не является побочным эффектом. Что же касается вторичной, то есть развившейся в процессе применения, толерантности, не преодолеваемой возможным увеличением дозы, то такое состояние часто оказывается крайне опасным. Так, толерантность к нитратам, развивающаяся у больных ИБС, чревата развитием острых коронарных эпизодов , как и внезапная отмена препаратов этой группы.
В зависимости от достоверности связи побочных реакций с используемым лекарственным средством побочные действия подразделяют на ряд категорий:
- Определенные побочные эффекты – возникающие на фоне применения препарата, купирующиеся после его отмены и вновь возникающие при возобновлении приема.
- Возможные побочные эффекты – возникающие на фоне применения препарата, купирующиеся после его отмены, но в связи с невозможностью повторного назначения (риск возникновения тяжелых осложнений или др.) не подтвержденные, что не позволяет отнести их к категории подтвержденных.
- Сомнительные побочные эффекты – возникающие на фоне применения препарата, но купирующиеся, несмотря на продолжение терапии.
По течению различают острые и хронические побочные эффекты.
В зависимости от тяжести клинических проявлений побочные эффекты могут быть разделены на:
- фатальные или смертельные (например, агранулоцитоз, развившийся при приеме хлорамфеникола [левомицетина], метамизола [анальгина, баралгина], сульфаниламидов);
- тяжелые, при которых очень высока опасность для жизни (анафилактический шок, желудочковая пароксизмальная тахикардия при лечении антиаритмическими средствами);
- средне-тяжелые, не несущие непосредственной угрозы жизни, но требующие не только отмены препарата, но и проведения специальной терапии;
- легкие, купирующиеся при отмене препарата или даже только при снижении дозы.
В З А И М О Д Е Й С Т В И Е Л Е К А Р С Т В
Возможность изменения фармакологического эффекта (за счет изменения фармакокинетики или фармакодинамики) лекарственного средства при одновременном или последовательном его назначении с другим препаратом хорошо известно и широко применяется для усиления лечебного действия или ослабления неблагоприятных реакций. Однако взаимодействие лекарств опасно возможностью потери терапевтического эффекта, увеличением частоты или степени выраженности известных побочных реакций или появлением новых, ранее не возникавших побочных эффектов.
В современном понимании термин взаимодействие лекарств подразумевает неблагоприятные последствия совместного использования различных препаратов, однако .часто он трактуется по-прежнему широко, в связи с чем продолжают выделять «полезное» и «вредное» взаимодействие.
Клиническое значение взаимодействия лекарств можно свести к нескольким положениям:
|
а) зависимость между дозой и эффектом и б) небольшая терапевтическая широта (сердечные гликозиды, антиаритмики, симпатомиметики, непрямые антикоагулянты, оральные сахароснижающие средства, гипотензивные, противоэпилептические препараты и др); |
|
а) вероятность развития клинически значимого взаимодействия увеличивается, если лекарства назначаются по одному и тому же поводу (комбинация дигоксина и хинидина при аритмиях), б) то или иное клинически значимое взаимодействие может возникнуть у одного больного и не проявиться у другого; |
|
Это требует знания фармакологических основ их взаимодействия и тщательного изучения с этой точки зрения литературы по каждому новому препарату |
В зависимости от механизмов выделяют следующие типы взаимодействия:
- фармацевтическое,
- фармакокинетическое,
- фармакодинамическое,
- фармакологическое (или физиологическое).
ФАРМАЦЕВТИЧЕСКОЕ ВЗАИМОДЕЙСТВИЕ
При возникновении химических или физико-химических реакций между лекарственными средствами вне человеческого организма («в шприце», инфузионном растворе, микстуре) говорят о фармацевтическом взаимодействии. В результате может выпадать осадок, появляться новых цвет, другой запах и т. п. Однако, как правило, внешний вид смеси не меняется, а теряются фармакологические свойства лекарств.
Фармацевтическое взаимодействие сегодня имеет значение почти исключительно при парентеральных путях введения, поскольку сложные прописи, применявшиеся в медицине ранее (микстуры, порошки и т. п.) практически повсеместно (за исключением, может быть, дерматологии, где широко используются сложные мази и «болтушки») заменены официнальными препаратами, в которых не допускается случайных сочетаний действующих веществ, а также веществ, составляющих лекарственную форму.
Взаимодействие «в шприце» определяется, прежде всего, рН смешиваемых растворов (взаимная нейтрализация веществ с кислой и щелочной реакциями или изменения оптимального рН, что приводит к изменению химических или физико-химических свойств одного из средств), а также окислительно-восстановительными реакциями. В качестве примера можно привести взаимодействие витаминов: в присутствии продуктов распада тиамина, всегда имеющихся в растворе, инактивируются другие витамины этой группы; тиамин несовместим с окисляющими и восстенавливающими веществами, карбонатами, ацетатами, рибофлавином, бензилпенициллином рибофлавином, глюкозой; цианокобаламин разрушается рибофлавином, особенно на свету, никотинамид ускоряет его фотолиз. В одном растворе сердечные гликозиды не совместимы с аминофиллином [эуфиллином](!), концентрированой глюкозой, дифенгидрамином [димедролом], тримеперидином [промедолом]. Инсулины различной длительности нередко вводятся одним шприцом, однако, например, протамин-цинк-инсулин содержит избыток протамина, способного дополнительно связывать корткодействующий инсулин. Гепарин быстро теряет активность в растворе глюкозы и в присутствии гидрокортизона, а норадреналин быстро окисляется в изотоническом растворе хлорида натрия. Необходимо подчеркнуть, что с препаратом может взаимодействовать не активное начало другого средства, а растворитель, стабилизатор, консервант и др. Так, тиамин полностью распадается в растворах, содержащих сульфиты, в связи с чем несовместим с раствором допамина, содержащим дисульфит.
Кроме того, некоторые вещества способны за счет физико-химического взаимодействия связывать лекарственные вещества., препятствуя созданию их терапевтических концентраций в крови. В первую очередь к ним относятся коллоидные растворы (декстран [реополиглюкин, полиглюкин], пентастарч [инфукол, рефортан], поливидон [гемодез]), препараты крови, рстворы аминокислот, жировые эмульсии, которые недопустимо использовать как среду для приготовления инфузионных растворов.
Для развития фармацевтического взаимодействия очень важен фактор времени. Поэтому смеси необходимо готовить непосредственно перед использованием и особое внимание обращать на инфузионные растворы, в которых может произойти взаимодействие, не успевающее проявиться при выполнении инъекций.
ФАРМАКОКИНЕТИЧЕСКОЕ ВЗАИМОДЕЙСТВИЕ
Если одно лекарство способно изменить фармакокинетические параметры другого, в результате чего меняются его эффекты, то говорят фармакокинетическом взаимодействии. В соответствии с этапами фармакокинетического цикла можно выделить и разные варианты фармакокинетического взаимодействия.
А. Взаимодействие при поступлении лекарств в организм (всасывание)
Обычно взаимодействие на этом этапе рассматривается как результат изменения всасывания лекарств в ЖКТ. Однако необходимо подчеркнуть, что и при парентеральном введении одни лекарства могут влиять на поступление других, если включается механизм всасывания (например, при внутримышечном или подкожном введении). Так, все средства, способные вызвать централизацию кровообращения и нарушение тканевой перфузии (катехоламины и их синтетические аналоги), будут ухудшать процессы адсорбции, где бы они ни проходили. Наиболее же часто взаимодействие этого этапа наблюдается при энтеральном применении лекарств.
Изменение скорости всасывания или количества всосавшегося вещества может быть результатом:
- изменения рН желудочного сока под влиянием системных (растворимых) антацидов и противосекреторных средств способствуют усилению ионизации слабых кислот, снижению их липофильности и абсорбции (салицилаты, антикоагулянты, некоторые сульфонамиды, фенилбутазон [бутадион]);
- изменения двигательной активности ЖКТ
- при замедлении эвакуации из желудка средствами с холиноблокирующими свойствами (некоторые антидепрессанты), антигистаминными препаратами, опиоидными анальгетиками уменьшается всаывание леводопы, разрушающейся в желудке,
- увеличение скорости прохождения по кишечнику глюкокортикоидов, дигоксина при систематическом приеме слабительных средств уменьшает их биодоступность;
- прямое взаимодействие в ЖКТ с адсорбирующими средствами, ионообменными смолами, средствами, образующими на поверхности слизистой оболочки препятствующие всасыванию пленки, а также образование неактивных хелатных соединений или комплексонов – активированный уголь, белая глина (каолин), пектиновые вещества препятствуют всасыванию множества лекарств, в частности, дигоксина, линкомицина; холестирамин – сердечных гликозидов, тироксина; кальций, магний, аллюминй, железо – тетрациклина; все антациды – многих средств, в частности, хлорпромазина[аминазина], фенитоина[дифенина]; здесь же следует упомянуть вазелиновое масло;
- изменения кишечной флоры под влиянием антибактериальных средств приводит к подавлению ее ферментативной и синтетической функций; в связи с этим, например, снижается активность оральных контрацептивов, поскольку нарушается реактивация и повторное всасывание конъюгированных стероидов (см. «энтерогепатическая циркуляция» в разделе «Элиминация») или потенцируется действие непрямых антикоагулянтов, поскольку снижается синтез витамина К.
- индивидуальное влияние одних препаратов на абсорбцию других – всасывание аскорбиновой кислоты уменьшается при одновременном приеме оральных кнтрацептивов, а аскорбиновая кислота повышает всасывание пенициллина, железа..
Б. Взаимодействие на уровне связывания с белком
Взаимодействие на уровне связывания с белком протекает по типу конкуренции за белок, в результате чего уменьшается связанная фракция, и увеличивается свободная. Повышение концентрации прпарата в крови приводит к возрастанию его активности и возможности передозировки. Такое взаимодействие подчиняется ряду закономерностей:
- феномен конкуренции и вытеснения проявляется клинически только у тех препаратов, связь которых с белком составляет более 80-90%;
- даже при высокой степени связи с белком этот феномен клинически не проявляется у препаратов с большим объемом распределения;
- различные препараты обладают разной степенью сродства к многочисленными фрагментам молекулы белка, способным связываться с лекарственными веществами, поэтому только высокой степени связи с белком еще недостаточно для развития данного типа взаимодействия;
- повышение концентрации свободной фракции в большинстве случаев сопровождается усилением элиминации (биотрансформации и экскреции) и снижением концентрации до установления нового равновесия, в связи с чем возрастание клинической активности вытесненного препарата будет временным, а наиболее опасно назначение вытесняющего препарата прерывисто или в изменяющихся дозах..
В качестве примеров взаимодействия на уровне конкуренции за белок можно привести возрастание эффекта непрямых антикоагулянтов и оральных сахароснижающих средств при совместном с ними применении нестероидных противовоспалительных средств или сульфаниламидов.
В. Взаимодействие на уровне распределения
Такое взаимодействие осуществляется за счет влияния одного из препаратов на кровоснабжение и, соответственно, на доставку другого. Так, улучшение кровообращения у больных с сердечной недостаточностью с помощью кардиотонических средств или улучшение реологических свойств крови позволяет лекарствам попастьв недоступные ранее участки органов. И наоборот, препараты, способствующие централизации кровообращения, препятствуют распределению лекарств в ткани. Нарушать фазу распределения могут и средства, вызывающие перераспределение крови с развитием «синдрома обкрадывания», например, миотропные спазмолитики (папаверин, дротаверин [но-шпа]).
Г. Взаимодействие на уровне биотрансформации
Как уже указывалось выше, активность ферментов, осуществляющих биотрансформацию лекарств, может изменяться под влиянием внешних факторов, в том числе и самих лекарств. В результате этого одни средства могут влиять на активность преобразования других. Известно несколько сотен лекарственных веществ, способных угнетать или стимулировать биотрансформацию.
- Индукция ферментов. Многие лекарственные средства, служащие субстратом для метаболизирующих ферментов, могут повышать биосинтез последних (индукция фермента субстратом). В результате усиливается биотрансформация как самого средства, так и других лекарств, метаболизирующихся с участием данного фермента. В качестве индукторов могут выступать снотворные и седативные (барбитураты, включая комбинированные препараты [беллатаминал, валокордин и др.],противосудорожные (фенитоин [дофенин], примидон [гексамидин, мисолин]), транквилизаторы (диазепам [реланиум, седуксен], хлордиазепоксид [хлозепид, элениум], мепробамат), нейролептики (хлорпромазин [аминазин], трифлуоперазин [стелазин, трифтазин]), противовоспалительные (фенилбутазон [бутадион]), сахароснижающие (толбутамид [бутамид, орабет] и др. средства. Активными индукторами ферментных систем являются инсектициды, алколь, кофе.
Усиление биотрансформации при сочетании лекарств с индуцирующими ферменты средствами приводит к падению эффективности лечения и необходимости увеличивать дозы. При отмене же индукторов возрастает концентрация второго препарата и усиливается его эффект. Классический пример – взаимодействие непрямых антикоагулянтов и фенобарбитала. Показано, что 14% кровотечений при лечении антикоагулянтами связанос отменой лекарств, индуцирующих микросомальные ферменты печени.
Систематический прием индукторов ферментов – фенитоина [дифенина], барбитуратов, включая комбинированные препараты [беллатаминал, валокордин и др.], применение рифампицина могут снижать эффективность оральных контрацептивов, что чревато нежелательной беременностью.
- Ингибиция ферментов. Лекарственные средства, угнетающие ферментативные системы и таким образом влияющие на метаболизм других химических веществ, потенцируют их действие, если его интенсивность и продолжительность зависят от скорости метаболизма. Среди ингибирующих ферменты лекарств можно назвать наркотические анальгетики, некоторые антибиотики (актиномицин, эритромицин и ряд др. макролидов), отдельные блокаторы Н2-гистаминовых рецепторов. Необходимо отметить, что, индуцируя одни ферменты, некоторые вещества способны ингибировать другие; к таким средствам относится, например, фенилбутазон [бутадион], который способен потенцировать действие толбутамида [бутамида, орабета], непрямых антикоагулянтов, фенитоина [дифенина]. Известно почти универсальное ингибирующее действие Н2-блокатора I поколения циметидина [гистодила, тагамета], что вызывает повышение плазменной концентрации непрямых антикоагулянтов, фенитоина [дифенина], пропранолола [анаприлина, обзадана], хлордиазепоксида [хлозепида, элениума], диазепама [реланиума, седуксена], некоторых трициклических антидепрессантов, лидокаина, теофиллина (аминофиллина [эуфиллина]), метронидазола [трихопола], метилдопы [допегита]. Считалось, что этим недостатком не обладают Н2-блокаторы II поколения. Однако ранитидин [зантак, гистак, безацид] способен подавлять биотрансформацию глибенкламида [манинила], метопролола, нифедипина [коринфара], фенитоина [дифенина], дисульфирама [тетурама, эспераля], теофиллина (аминофиллина [эуфиллина]), оральных контрацептивов, а также этанола.
Реакции подобного типа бывают весьма тяжелыми. Так, известны случаи гипогликемических ком при назначении эритромицина на фоне приема толбутамида [бутамида, орабета]. Описаны летальные случаи при одновременном назначении азатиоприна или 6-меркаптопурина с аллопуринолом (ингибирующим фермент, принимающий участие в окислении этих цитостатиков) и циметидина с теофиллином (см. выше).
Биотрансформация этанола ингибируется метронидазолом [трихополом], оральными сахароснижающими препаратами из группы сульфонилмочевины, но в наибольшей степени – дисульфирамом [тетурамом, эспералем], что вызывает крайне неприятные ощущения, а при неадекватных дозах может вызвать интоксикацию.
Взаимодействие лекарств может происходить при изменении активности и других ферментов. Так, передозировка и тяжелые осложнения возникают при терапии катехоламинами пациентов, получающих ингибиторы моноаминоксидазы – фермента, участвующего в окислении катехоламинов (активностью ингибитора моноаминоксидазы обладает, например, фуразолидон).
Г. Взаимодействие на уровне выведения (экскреции)
Поскольку основным органом, осуществляющим экскрецию, являются почки, взаимодействие на уровне выведения в первую очередь происходит здесь.
- Взаимодействие во время пассивной диффузии происходит в связи с изменениями рН мочи. Так при ощелачивании мочи вследствие применения антацидных средств увеличивается экскреция слабых кислот (которые ионизируются, теряют липофильные свойства и в связи с этим не реабсорбируются) – салицилатов, барбитуратов, а их плазменная концентрация падает; экскреция же слабых оснований – эфедрина, хинидина снижается. Причем это снижение может вызываться как гидрокарбонатом, так и ацетазоламидом [диакарбом]. Закисление мочи хлоридом аммония, большими дозами аскорбиновой кислоты увеличивает экскрецию эфедрина и хинидина.
- Снижение почечного клиренса может возникать вследствие конкуренции за активную секрецию у препаратов, экскретирующихся таким путем. Так, салицилаты и сульфаниламиды тормозят экскрецию метотрексата, фуросемид снижает экскрецию гентамицина. При этом если в конкуренции участвует нефротоксический препарат, то его токсическое действие усиливается.
ФАРМАКОдинамическое ВЗАИМОДЕЙСТВИЕ
Фармакодинамическое взаимодействие осуществляется на уровне механизма действия лекарств.
- Взаимодействие на рецепторах, как правило, протекает по типу конкуренции за рецептор и нередко наблюдается при одновременном назначении агонистов и антагонистов: агониста бета2-адренорецепторов сальбутамола [вентолина] и бета-блокатора пропранолола [анаприлина, обзидана], агониста и антагониста опиатных рецепторов морфина и налоксона и т. п. Результат такого взаимодействия зависит от соотношения доз и от выраженности аффинитета каждого из участников процесса к рецептору. С практической точки зрения представляет интерес взаимодействие двух агонистов или двух антагонистов одного рецептора. Так, если на фоне слабого наркотического анальгетика, имеющего высокий аффинитет к опиатным рецепторам, кодеина вводится более сильный – морфин, то ожидаемое усиления клинических эффектов опиатов не возникнет, поскольку с занятыми кодеином рецепторами морфин взаимодействовать не может.
- Другой вариант взаимодействия на рецепторах – изменение их чувствительности к агонистам. Дефицит натрия ослабляет ответы адренорецепторов к катехоламинам, а дополнительное введение натрия – увеличивает. Эффективность глюкокортикоидов при бронхообструкции обусловлена увеличением количества бета2-адренорецепторов бронхов (то есть увеличение рецепторного поля к влиянию бета2-агонистов) однако аналогичное их действие на другие органы, например, сердце, усугубляет побочные эффекты (тахикардия). Повышение чувствительности бета-адренорецепторов к катехоламинам вызывает и средство для ингаляционного наркоза фторотан (угроза фибрилляции при введении бета-стимуляторов – адреналина, норадреналина и др.)
- Препараты, влияющие на ферменты, также могут вступать в лекарственные взаимодействия. Так, ингибитор протеаз апротинин [гордокс, контрикал, трасилол] подавляет и активность неспецифической сывороточной холинэстеразы, что у лиц с небольшими запасами этого фермента (на нижней границе нормы) может удлинить действие деполяризующего миорелаксанта суксаметония [дитилин, листенон] за счет накопления ацетилхолина, усугубляющего деполяризацию. Ингибиция протеаз может препятствовать проведению фибринолитической терапии стрептокиназой, урокиназой или t-РА.
ФАРМАКОЛОГическое (ФИЗИОЛОГИЧЕСКОЕ) ВЗАИМОДЕЙСТВИЕ
Этот тип взаимодействия осуществляется на уровне эффекта и, вероятно, встречается наиболее часто. Заключается физиологическое взаимодействие в том, что в результате фармакологического эффекта оного препарата так изменяются какие-либо функции, что действие другого лекарственного средства проявляется неожиданно.
Классический пример такого взаимодействия – нарушения сердечного ритма, вызванные сердечными гликозидами на фоне электролитных нарушений, возникших в результате применения диуретиков. Вероятность гликозидной интоксикации усиливается при дополнительном назначении препаратов кальция, концентрация которого в цитоплазме кардиомиоцитов увеличивается под влиянием гликозидов. По этой же причине опасной комбинацией является сочетание сердечных гликозидов и блокатора кальциевых каналов верапамила [изоптина, финоптина], нарушающего кальциевые токи и способствующего в этих условиях перегреске клеток кальцием. Антибактериальные препараты способны потенцировать действие непрямых антикоагулянтов вплоть до развития кровотечений; этот эффект опосредуется через нарушение синтеза витамина К флорой кишечника вследствие ее подавления.
Нерациональные сочетания:
- антикоагулянты с антиагрегантами – увеличение риска геморрагических осложнений,
- антикоагулянты с нестероидными противовоспалительными средствами – увеличение риска желудочно-кишечных кровотечений,
- нестероидные противовоспалительные средства с глюкокортикоидами – увеличение риска ульцерогенного действия,
- глюкокортикоидов с диуретиками – увеличение риска электролитных нарушений и остеопороза,
- бета2-агонистов с теофиллином (аминофиллином [эуфиллином]) – увеличение риска сердечных аритмий
и т. д., и т. п.
Крайне опасно внутривенное введение верапамила [изоптина, финоптина] пациентам, получающим пропранолол внутрь; возникающая при этом депрессия функций сердца чревата летальным исходом.
В заключение следует подчеркнуть, что взаимодействие лекарственных средств – один из самых коварных феноменов в фармакотерапии. Часто оно возникает непредсказуемо, его проявления неожиданны, а результаты иногда бывают фатальны. Важно обратить внимание еще и на то, что частое развитие взаимодействия лекарств прямой результат полипрагмазии. Так, например, у пациентов, одновременно получающих 6 и более препаратов осложнения терапии, связанные с взаимодействием лекарств, развиваются в 7 раз чаще, чем при меньшем количестве лекарственных средств.