



Синдром Бругада: клеточные механизмы и подходы к лечению

Внезапная сердечная смерть (ВСС)
является одной из ведущих причин смертности в развитых странах. Частота ее развития составляет 15–20% от всех ненасильственных случаев смерти [2].
К заболеваниям, ассоциированным с высоким риском ВСС в молодом возрасте, относят синдром внезапной смерти младенцев (sudden infant death syndrome), синдром удлиненного интервала Q–T (СУИ Q–T), синдром внезапной необъяснимой смерти (sudden unexplained death syndrome), аритмогенную дисплазию правого желудочка, идиопатическую фибрилляцию желудочков и др. Одно из наиболее загадочных заболеваний в этом ряду – синдром Бругада (CБ). Несмотря на то что во всем мире опубликованы сотни работ, посвященных данному заболеванию, на крупнейших международных кардиологических конгрессах регулярно проводятся соответствующие тематические секции, в отечественной литературе имеются лишь единичные описания синдрома, далеко не всегда полностью отражающие типичную картину заболевания. СБ является, по мнению многих специалистов, «ответственным» за более чем 50% внезапных некоронарогенных смертей в молодом возрасте.
В 1992 г. братья Р. и J. Brugada [16] (рис. 1) опубликовали результаты наблюдения за 8 пациентами (6 мужчин и 2 женщины), имевшими в анамнезе синкопальные состояния и эпизоды клинической смерти. Для верификации их причин пациентам были выполнены инструментальные исследования: электро-, эхокардиография, нагрузочное тестирование, ангиография, электрофизиологическое исследование (ЭФИ). У 4 пациентов была проведена биопсия миокарда. Выполненные исследования не позволили выявить какое-либо органическое заболевание сердца. Единственной особенностью у наблюдаемых пациентов было на-личие блокады правой ножки пучка Гиса (БПНПГ) и подъема сегмента ST в правых грудных отведениях на ЭКГ покоя.

Рис. 1. Братья Бругада: слева направо Рамон, Педро, Джозеф.
Во время синкопальных состояний у них регистрировалась полиморфная желудочковая тахикардия (ЖТ), которая возникала после ранних парных желудочковых экстрасистол и легко индуцировалась с помощью программированной стимуляции. Впоследствии 4 больным имплантировали кардиовертер-дефибриллятор (ИКВД), еще у 2 нарушения ритма исчезли на фоне приема b-блокаторов. У седьмого пациента аритмия контролировалась за счет приема амиодарона и дифенилгидантоина на фоне электрической стимуляции сердца в режиме VVI. Восьмой пациент, принимавший амиодарон, неожиданно умер во время имплантации кардиостимулятора. Так братьями Бругада впервые были описаны характерные изменения на электрокардиограмме, позволяющие определить высокий риск возникновения желудочковых нарушений ритма у лиц без органических изменений сердца.
В 93,3% случаев приступы ЖТ при СБ возникают в вечернее и ночное время (с 18 до 06 ч), чаще во второй половине ночи, что подтверждает роль усиления вагусных влияний в возникновении фибрилляции желудочков (ФЖ) при СБ. Этот циркадный паттерн свидетельствует также о различиях в патогенезе возникновения фатальных аритмий у паци-ентов с СБ и ишемической болезнью сердца, когда основной циркадный пик ВСС приходится на ран-ние утренние часы (Deedwania P., 1998).
Необходимо проводить дифференциальную диагностику СБ и ряда заболеваний, которые могут стать причиной схожих ЭКГ-проявлений: аритмогенной дисплазии правого желудочка (ПЖ), миокардита, кардиомиопатии, болезни (миокардита) Чагаса (Chagas), болезни Штейнертa, опухоли средостения. У всех больных с синкопальными состояниями неясной этиологии, ночными пароксизмами уду-шья, случаями ВСС в семье (особенно в молодом возрасте и в ночное время), типичным ЭКГ-паттерном необходимо исключить СБ. Для этого таким пациентам следует проводить фармакологические пробы, динамическое ЭКГ-обследование (в том числе родственников больного), холтеровское мониторирование (ХМ). Одним из наиболее до-стоверных методов диагностики СБ является молекулярно-генетическое исследование.
К патологиям, вызывающим элевацию сегмента ST в правых грудных отведениях, относятся: острый миокардит, острый перикардит, гемоперикард, инфаркт правого желудочка, диссекция аневризмы аорты, острая тромбоэмболия легочной артерии, аномалии центральной нервной системы, мышечная дистрофия Дюшенна, атаксия Фридрейха, недостаточность тиамина, гиперкальциемия, гиперкалиемия, mediastinal tumor compresing right ventricular outflow tract, аритмогенная кардиомиопатия правого желудочка, СУИ Q–T 3 тип, блокада правой ножки пучка Гиса, блокада левой ножки пучка Гиса, гипертрофия левого желудочка, синд-ром ранней реполяризации, гипотермия.
Кроме того, ЭКГ-изменения, сходные с отмечаемыми при СБ, способны вызвать следующие лекарственные и другие средства:
1. Антиаритмические препараты:
а) блокаторы натриевых каналов:
– класс IC (флекаинид, пропафенон, пилсикаинид),
– класс IA (аймалин, прокаинамид, дизопирамид);
б) блокаторы кальциевых каналов:
– верапамил.
2. Бета-блокаторы:
пропранолол и т. п.
3. Антиангинальные препараты:
а) блокаторы кальциевых каналов:
– нифедипин, дилтиазем;
б) нитраты:
– изосорбид динитрат, нитроглицерин;
в) активаторы калиевых каналов: – никорандил.
4. Психотропные препараты:
а) трициклические антидепрессанты:
– амитриплин, – нортриптилин, – дезипрамин, – кломипрамин;
б) тетрациклические антидепрессанты:
– мапротилин;
в) фенотиазин: – циамемазин, – перфеназин;
г) селективный ингибитор обратного захвата серотонина:
– флуоксетин.
5. Другие препараты:
– дименгидрин,
- – интоксикация кокаином,
- – алкогольная интоксикация.
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА СИНДРОМА БРУГАДА
Синдром Бругада представляет собой генетически детерминированное нарушение сердечного ритма, характеризующееся синкопальными состояниями, подъемами сегмента ST (с точки J) над изоэлектрической линией в правых прекардиальных отведениях (V1–V3), которые могут транзиторно исчезать, также в этих отведениях могут регистрироваться инвертированный зубец Т, полная или неполная БПНПГ, периодическое удлинение интервала P–R с высоким риском развития жизнеугрожающих желудочковых тахиаритмий (эпизодов полиморфной ЖТ и ФЖ), обычно развивающихся во сне или в спокойном состоянии. Нередко у больных обнаруживаются также наджелудочковые аритмии, чаще фибрилляция предсердий (ФП). В большинстве случаев СБ наблюдается у пациентов в 30–40-летнем возрасте. Впервые данный синдром был описан у трехлетней девочки, имевшей частые эпизоды потери сознания и впоследствии внезапно погибшей, несмотря на активную антиаритмическую терапию и имплант кардиостимулятора. Клиническая картина заболевания характеризуется частым возникновением синкопе на фоне приступов ЖТ и ВСС, преимущественно во сне, а также отсутствием признаков органического поражения миокарда при аутопсии.
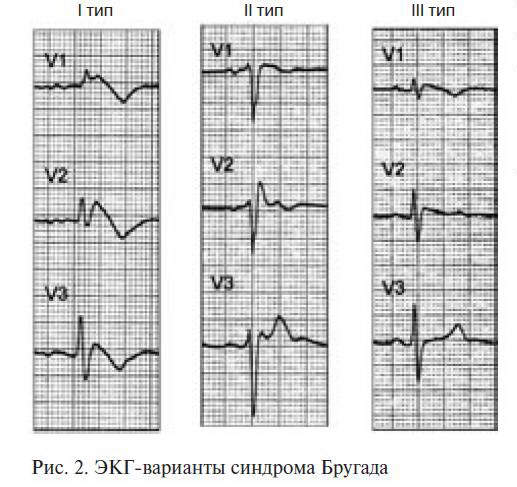
Выделяются следующие клинико-электрокардиографические формы и варианты СБ [3] (табл. 1, рис. 2):
– полная форма (типичная ЭКГ-картина с синкопе, предсинкопе, случаями клинической смерти или ВСС вследствие полиморфной ЖТ);
– типичная ЭКГ-картина у асимптоматичных больных без семейной истории ВСС или СБ;
– типичная ЭКГ-картина у асимптоматичных больных, членов семей больных с полной формой синдрома;
– типичная ЭКГ-картина после проведения фармакологических тестов у асимптоматичных обследуемых, членов семей больных с полной формой синдрома;
– типичная ЭКГ-картина после проведения фармакологических тестову больных с повторными синкопе или идиопатической ФП;
– типичная ЭКГ-картина с явной БПНПГ, элевацией сегмента ST и удлинением интервала P–R;
– типичная ЭКГ-картина с элевацией сегмента ST, но без удлинения интервала P–R и БПНПГ;
– неполная БПНПГ с умеренной элевацией сегмента ST;
– изолированное удлинение интервала P–R. Выраженность клинических проявлений СБ определяется степенью повреждения натриевых каналов: при повреждении менее 25% каналов ЭКГ-паттерн и нарушения ритма наблюдаются только после введения блокаторов натриевых ка-налов, при увеличении количества поврежденных натриевых каналов (свыше 25%) резко возрастает риск ВСС [43].
Та б л и ц а 1. Диагностические критерии синдрома Бругада. Аномалии сегмента ST в V1–V3 отведениях
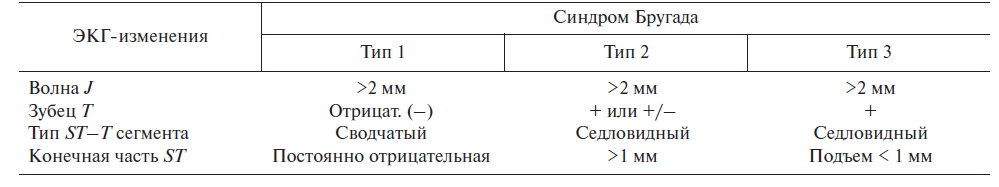

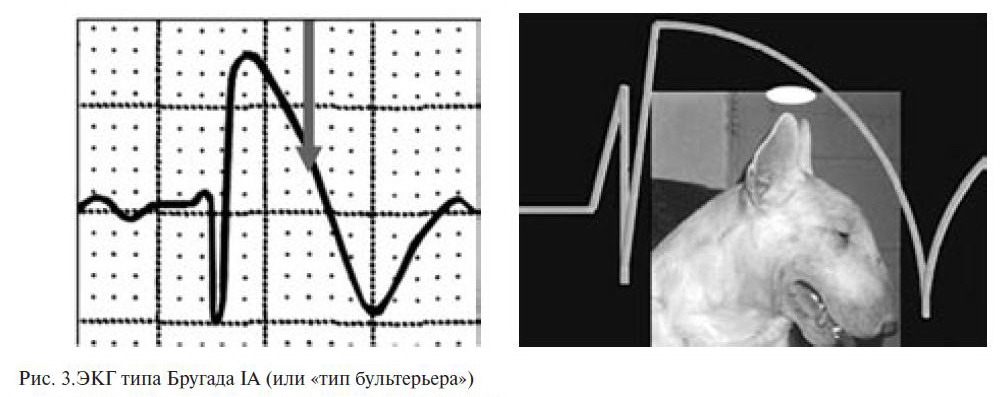
Описаны два типа подъема сегмента ST при СБ:
«saddle-back type» («седло») и «јoved type» («свод») (рис. 3).
Подъем «јoved type» достоверно преобладает при симптомных формах СБ, с torsades de pointes (TdP; пируэтная желудочковая тахикардия) и ФЖ в анамнезе, в то время как «saddle-back type» чаще встречается при бессимптомных формах СБ.
Однако изменения на ЭКГ при СБ могут носить преходящий характер, что требует поиска дополнительных методов верификации. Некоторые авторы для диагностики СБ предлагают использовать высокие (на 1–2 межреберья выше) правые грудные отведения.
Для верификации диагноза необходимо проведение нагрузочных проб с лекарственной нагрузкой (антиаритмические препараты IA класса), с введением блокаторов натриевых каналов (аймалин – 1 мг/кг за 5 мин внутривенно или 400 мг перорально, новокаинамид – 10 мг/кг за 10 мин внутривенно, пилсикаинид – 1 мг/кг за 10 мин внутривенно) [6].
Тест с блокаторами натриевых каналов проводится в условиях реанимационного отделения, так как во время его проведения существует большая вероятность развития ЖТ по типу TdP и ФЖ.
Надо отметить, что полиморфная ЖТ больше ассоциирована с СБ, тогда как мономорфная ЖТ встречается реже и в основном распространена у детей [44].

ПРОГНОЗ И СТРАТИФИКАЦИЯ РИСКА
Частота встречаемости СБ более низкая в западных странах (1–2 случая на 10 000 человек) и повышается в Юго-Восточной Азии (более 5 на 10 000), особенно в Таиланде и на Филиппинах. Средний возраст больных на момент постановки диагноза составляет 40±22 года. Типичные для синдрома Бругада изменения на ЭКГ начинают проявляться лишь после 5-летнего возраста [38].
Главная цель стратификации – прогноз потенциального риска возникновения эпизодов желудочковых аритмий. Стратификация проводится на основании клинических характеристик больных, указанных в Руководстве по ведению больных с ЖТ и профилактике ВСС (ACC/AHA/ESC, 2006): На данный момент четких данных, связывающих положительный семейный анамнез с аритмическими осложнениями, нет, поэтому не следует относить всех членов семьи пациента с СБ к группе повышенного риска.
Существует общепринятое мнение о том, что синкопальные состояния являются важным предиктором возникновения жизнеугрожающих аритмий. Однако не всегда удается поставить точный этиологический диагноз, так как у молодых людей часто имеют место вазовагальные синкопе. Спонтанные изменения на электрокардиограмме («сводчатые» или «седловидные» сегменты ST) позволяют выявить группу пациентов с более высоким риском возникновения жизнеугрожающих аритмий. Часто спонтанные изменения на электрокардиограмме могут носить интермиттирующий характер.
Так, P. Brugada и соавт. обнаружили, что воспроизводимость ЭКГ-паттерна СБ при повторных записях ЭКГ составляет лишь 25%.
Важно отметить, что пациенты с синкопе в сочетании со спонтанными изменениями на электрокардиограмме имеют риск возникновения ЖТ в 6 раз выше, чем у пациентов со спонтанными изменениями на электрокардиограмме, но без синкопе. Как указывает R. Brugada, холтеровское мониторирование у пациентов с СБ может показать либо отсутствие аритмий, либо желудочковую аритмию, приводящую к возникновению у больного ВСС.
По мнению Л. М. Макарова, такие данные ХМ, как укорочение интервала Q–T, наличие паузозависимых правожелудочковых тахиаритмий в ночное время, высокие значения циркадного индекса (более 1,5) и увеличение rMSSD, могут помочь в диагностике СБ.
Что касается генетических мутаций, то сами по себе мутации в гене SCN5A не определяют группу пациентов с высоким аритмическим рис-ком. Однако ранняя идентификация бессимптомных носителей СБ может предотвратить ВСС.
Роль ЭФИ в стратификации риска при СБ окончательно не установлена. По мнению братьев Brugada, ЭФИ играет важную роль, при этом наличие положительного прогностического результата составляет 23%, а отрицательного – 77%.
В то же время S. Priori и соавт. предлагают использовать неинвазивные методы стратификации риска, основанные на изменениях электрокардиограммы и клинических симптомах.
Так как СБ может проявляться в любом возрасте, необходимо исключить его в следующих случаях:
– появление характерных изменений на электрокардиограмме;
– синкопальные состояния (обмороки) неясного происхождения;
– эпизоды полиморфной ЖТ;
– случаи внезапной смерти в семье, особенно у практически здоровых мужчин в возрасте 30–50 лет.
У мужчин СБ встречается в 8–10 раз чаще, чем у женщин [24]. Необходимо принять во внимание, что большинство пациентов в исследованиях, по священных изучению СБ, – мужчины. При этом в связи с небольшим числом женщин среди пациентов нет четких данных об отличительных характеристиках течения синдрома у женщин [17]. В некоторых странах Юго-Восточной Азии этот синдром является одной из наиболее частых при-чин смерти среди молодых мужчин – 4–10 случаев ВСС на 10 000 населения в год. Существует даже мнение, что до 50% случаев ВСС среди пациентов без органических поражений сердца в этом регионе может быть вызвано СБ [14].
Провоцирующими факторами развития СБ в этом регионе можно считать содержание большого количества калия в пище и жаркий климат. Исследования по данному вопросу продолжаются. Семейные случаи ВСС считают фактором риска развития ВСС. Однако в отличие от синдрома удлиненного интервала Q–T нет данных, которые бы подтверждали прогностическое значение отягощенного семейного анамнеза. В связи с этим предположение о том, что отсутствие отягощенного семейного анамнеза указывает на низкий риск ВСС, может быть ошибочным [1].
Брадикардия (из-за симпатовагального дисбаланса или других факторов) может способствовать возникновению аритмии. В исследовании аномальный метаиодбензилгуанид (MIBG) встречался у 8 (17%) из 17 пациентов с СБ, но ни у одного пациента контрольной группы не обнаружено уменьшения MIBG в нижней и перегородочн стенке ЛЖ, где была выявлена симпатическая дисфункция. Изучение MIBG в ПЖ, особенно в его выводном тракте (ВТ), затруднено. Кроме того, остается неясным, какую роль играет в формировании аритмии при СБ уменьшение MIBG. Если ВТ ПЖ также затронут, то этот дефект может действительно изменить симпатовагальный баланс в пользу развития аритмогенных субстратов.

Одним из факторов риска развития ВСС при СБ некоторые авторы считают увеличение площади поверхности тела, с которой регистрирует подъем сегмента SТ при поверхностном картировании ЭКГ, в частности, у больных, у которых при программированной желудочковой стимуляции была индуцирована ЖТ, этот показатель составил 183±139 см2 по сравнению с 52±58 см2 у больных при отсутствии индукции (для площади 50 см2 и более позитивный, и негативный предикторные уровни составили 92 и 60% соответственно). В этом исследовании наличие поздних потенциалов также коррелировало с индукцией ЖТ (позитив-ный и негативный предикторные уровни 100 и 75% соответственно).
ГЕНЕТИЧЕСКИЕ ОСНОВЫ СИНДРОМА БРУГАДА
СБ наследуется по аутосомно-доминантному типу и связан с мутациями в гене SCN5A, расположенном в коротком плече 3-й хромосомы 3р21-24, кодирующей субъединицу альфа-натриевых ка-налов кардиомиоцитов. Этот ген кодирует структуру белка a-субъединицы натриевых каналов, обеспечивающих натриевый ток потенциала действия (ПД). Мутации в этом гене также могут приводить к СУИ Q–T и нарушениям сердечной проводимости. Данная мутация считается первичной «электрической» болезнью сердца, развивающейся вследствие аномальной электрофизиологической активности эпикарда ПЖ в области выносящего тракта. Некоторое количество мутаций этого гена может вызывать ряд синдромов, которые накладываются друг на друга. Совсем недавно C. Antzelevitch и соавт. открыли два новых гена, вызывающих элевацию сегмента ST и укорочение интервала Q–T, что приводит к комбинации СБ с синдромом короткого интервала Q–T (СКИ Q–T) [42].
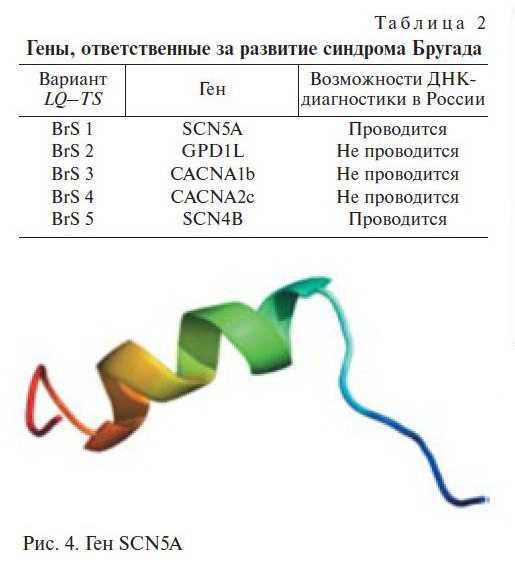
На сегодняшний день известны, по крайней мере, пять генов, ответственных за развитие СБ. Мутация в любом из них может привести к разви-тию заболевания (табл. 2).
При анализе гена SCN5A удается выявить мутации и установить молекулярно-генетическую форму заболевания у 20% пробандов (рис. 4) [28].
ЭЛЕКТРОФИЗИОЛОГИЧЕСКИЕ ОСНОВЫ СИНДРОМА БРУГАДА
Ионным механизмом СБ при мутации SCN5A является уменьшение количества или ускоренная инактивация натриевых каналов в клетках эпикар-да ПЖ [22], что приводит к уменьшению плотности потока натрия и преждевременной реполяризации эпикарда. Кроме того, при этом синдроме было обнаружено перемещение натриевых каналов с поверхности клеток в эндоплазматический ретикулум, что также нарушает их функцию [30]. Увеличение калиевого тока могло бы иметь подобный эффект, однако этот механизм у больных с СБ до настоящего времени не описан. Потеря вершины ПД на некоторых участках эпикарда, при его нормальной величине в эндокарде, создает дисперсию реполяризации стенки желудочка, приводящую к трансмуральному градиенту напряжения, который проявляется на ЭКГ подъемом сегмента ST. Вероятно, вследствие указанных выше процессов образуется «уязвимое окно», во время которого мо-жет возникнуть механизм reentry, запускающий ЖТ и ФЖ. Таким образом, мутация в гене приводит к потере функции каналов, что создает гетерогенность рефрактерных периодов – идеальный субстрат для механизма reentry и желудочковых аритмий. Возникновению ЖТ и ФЖ у таких больных, как правило, предшествует преждевременное сокращение желудочков с интервалом сцеп-ления 388±28 мс [12].
При записи электрокардиограммы непосредственно от эпи- и эндокарда ПЖ у больных с СБ с так называемым coved type подъемом сегмента ST от эпикарда был получен ПД в форме «spike-and-dome» («шип и купол»), а ПД от эндокарда не отличался от полученного в контрольной группе, что служило проявлением трансмурального градиента напряжения. Конфигурация «шип и купол» также может удлинять ПД эпикарда и способствовать быстрой реверсии трансмуральных градиентов, что приводит к записи на электрокардиограмме инвертированного зубца Т [18]. Другие авторы считают, что причиной подъема сегмента ST служит парадоксальное сокращение реполяризации в эпикарде ВТ ПЖ.
Получены данные, доказывающие стимулирующее влияние вагуса на развитие ФЖ. В этом исследовании у 6 пациентов на фоне СБ эпизоды ФЖ имели место во время сна, отдыха или мочеиспускания, то есть в моменты замедления синусового ритма и возрастания значения HF (высокочастотной составляющей спектра ритмограммы), и не возникали во время физической нагрузки. На ЭКГ признаки СБ появлялись после введения пропранолола, гипервентиляции и, наоборот, исчезали после физической нагрузки и инфузии изопротеренола. Было показано, что активация парасимпатической или торможение симпатической нервной системы играет роль в аритмогенезе при СБ. Об этом же свидетельствует увеличение подъема сегмента ST при введении холиномиметиков.
Доказательством симпатической дисфункции при СБ служит также исследование G. Baroudi и соавт. [11], в котором было обнаружено местное снижение обратного захвата I-MIBG (аналога входящего натриевого тока ведет к изменениям во 2-й фазе трансмембранного ПД в клетках эпикарда выводного тракта ПЖ. Это, в свою очередь, приводит к асинхронной реполяризации и электрической нестабильности в этом отделе сердца. Основными факторами аритмической «уязвимости» миокарда являются: парасимпатические влияния (сон, погружение в холодную воду, ныряние, прием пищи), повышение температуры тела, блокада натриевых и кальциевых каналов, ишемия миокарда и др. В этих условиях может генерироваться механизм повторного входа во 2-й фазе ПД с последующим возникновением желудочковых аритмий. В то же время у пациентов с СБ наблюдается нормализация сегмента ST после введения бета-адренергических стимуляторов или ингибиторов фосфодиэстеразы III. В недавнем исследовании J. Brugada и соавт. было показано, что степень элевации сегмента ST в отведении V1 является независимым фактором риска для ВСС или возникновения ФЖ.
Далеко не всегда СБ сопровождается выявле-нием при генетическом анализе мутации в SCN5A. В связи с этим предполагается, что СБ может быть вызван и другими мутациями, которые еще не идентифицированы. Имеется описание бессимптомного варианта указанного синдрома при мутации, приводящей к нарушению связи кальмодулина с натриевым каналом, что вызывает нарушение модуляции работы натриевого канала ионами кальция.
Исследователи предполагают [35], что кроме генетических нарушений важную роль в развитии полиморфной ЖТ и ФЖ при СБ играет активность автономной нервной системы. T. Kurita и соавт. субстрат для механизма reentry и желудочковых аритмий. Возникновению ЖТ и ФЖ у таких боль-ных, как правило, предшествует преждевремен-ное сокращение желудочков с интервалом сцеп-ления 388±28 мс [12].
При записи электрокардиограммы непосредственно от эпи- и эндокарда ПЖ у больных с СБ с так называемым coved type подъемом сегмента ST от эпикарда был получен ПД в форме «spike-and-dome» («шип и купол»), а ПД от эндокарда не отличался от полученного в контрольной группе, что служило проявлением трансмурального градиента напряжения. Конфигурация «шип и купол» также может удлинять ПД эпикарда и способствовать быстрой реверсии трансмуральных градиентов, что приводит к записи на электрокардиограмме инвертированного зубца Т [18]. Другие авторы считают, что причиной подъема сегмента ST служит парадоксальное сокращение реполяризации в эпикарде ВТ ПЖ. Генетически детерминированное угнетение входящего натриевого тока ведет к изменениям во 2-й фазе трансмембранного ПД в клетках эпикарда выводного тракта ПЖ. Это, в свою очередь, приводит к асинхронной реполяризации и электрической нестабильности в этом отделе сердца. Основными факторами аритмической «уязвимости» миокарда являются: парасимпатические влияния (сон, погружение в холодную воду, ныряние, прием пищи), повышение температуры тела, блокада натриевых и кальциевых каналов, ишемия миокарда и др. В этих условиях может генерироваться механизм повторного входа во 2-й фазе ПД с последующим возникновением желудочковых аритмий. В то же время у пациентов с СБ наблюдается нормализация сегмента ST после введения бета-адренергических стимуляторов или ингибиторов фосфодиэстеразы III. В недавнем исследовании J. Brugada и соавт. было показано, что степень элевации сегмента ST в отведении V1 является не-зависимым фактором риска для ВСС или возникновения ФЖ.
Далеко не всегда СБ сопровождается выявлением при генетическом анализе мутации в SCN5A. В связи с этим предполагается, что СБ может быть вызван и другими мутациями, которые еще не идентифицированы. Имеется описание бессимптомного варианта указанного синдрома при мутации, приводящей к нарушению связи кальмодулина с натриевым каналом, что вызывает нарушение модуляции работы натриевого канала ионами кальция.
Исследователи предполагают [35], что кроме генетических нарушений важную роль в развитии полиморфной ЖТ и ФЖ при СБ играет активность автономной нервной системы. T. Kurita и соавт получены данные, доказывающие стимулирующее влияние вагуса на развитие ФЖ. В этом исследова-нии у 6 пациентов на фоне СБ эпизоды ФЖ имели место во время сна, отдыха или мочеиспускания, то есть в моменты замедления синусового ритма и возрастания значения HF (высокочастотной составляющей спектра ритмограммы), и не возникали во время физической нагрузки. На ЭКГ признаки СБ появлялись после введения пропранолола, гипервентиляции и, наоборот, исчезали после физической нагрузки и инфузии изопротеренола. Было показано, что активация парасимпатической или торможение симпатической нервной системы играет роль в аритмогенезе при СБ. Об этом же свидетельствует увеличение подъема сегмента ST при введении холиномиметиков.
Доказательством симпатической дисфункции при СБ служит также исследование G. Baroudi и соавт. [11], в котором было обнаружено местное снижение обратного захвата I-MIBG (аналога норадреналина) пресинаптической мембраной кардиальных нейронов у 47% пациентов с СБ, и такого снижения не отмечено ни у одного обследованного контрольной группы. Показано также, что катехоламины уменьшают подъем сегмента ST и в ряде случаев купируют ФЖ.
В патогенезе СБ, возможно, играет роль дисба-ланс между a- и b-адренергической стимуляцией. При введении пациентам a-адреномиметиков или b-адреноблокаторов наблюдалось увеличение подъема сегмента ST, а при введении a-адреноблокаторов и b-адреномиметиков – его уменьше-ние [33].
Необходимо исследование пре- и постсинаптической функции симпатического и парасимпатического отделов вегетативной нервной системы для решения вопроса, является ли СБ первичной симпатической дисфункцией или дисбалансом между этими двумя отделами.
За последние несколько лет более 100 мутаций в гене SCN5A связывались с СБ. Лишь некоторые из этих мутаций были изучены в системах экспрессии и показана потеря функций по следующим механизмам:
1) нарушение в экспрессии натриевого канала;
2) изменения зависимости между вольтажом и временем активации, инактивация или реактивация натриевого тока (INa);
3) состояние промежуточной инактивации входа в натриевый канал с медленным восстановлением этого состояния;
4) ускоренная инактивация натриевого канала.
В системе экспрессии in vitro преждевременная инактивация натриевого канала иногда наблюдается при физиологических, но не при комнатных температурах [50]. С момента публикации этой информации было идентифицировано достаточно много пациентов с СБ, у которых под влиянием повышенной температуры тела развивалась полиморфная ЖТ [20].
Мутации в гене SCN5A являются ответственными за приблизительно 18–30% случаев СБ. Большинство указанных мутаций наблюдается при семейных формах синдрома. Отрицательный анализ SCN5A не исключает генетических нарушений, так как исследования промоторной зоны, мутации типа криптик сплайсинг или некоторые значительные генные перестройки не входят в обычный рутинный генетический анализ. В недавней публикации K. Hong и соавт. [31] впервые сообщяется о дисфункциональном натриевом канале в семье с СБ, возникшим вследствие интронной мутации, что влечет за собой активацию сайта криптик сплайсинг, нарушающего переработку mRNA в гене SCN5A. Делеция некоторых зон сегментов 2 и 3 IV домена SCN5A приводит к полной потере функции гена.
C. R. Bezzina и соавт. предложили некоторые доказательства гипотезы о том, что полиморфизм в промоторной зоне SCN5A – частое явление среди жителей Средней Азии – модулирует сердечную проводимость и может способствовать высокой частоте встречаемости СБ среди представителей азиатской расы [13]. Последовательность промотора SCN5A позволяет идентифицировать вариант гаплотипа, состоящего из 6 полиморфизмов с почти полным нарушением равновесия сцепления. Этот гаплотип встречается с аллельной частотой 22% у жителей Средней Азии и не наблюдается в контрольной группе (среди европейцев и афроамериканцев).
Был найден также второй локус, в 3-й хромосоме, близко расположенный, но отличающийся от локуса, где находится ген SCN5A [31]. Найденный локус показал генеалогическую корреляцию, при этом синдром был связан с прогрессирующими нарушениями проводимости, низкой чувствительностью к прокаинамиду и характеризовался относительно хорошим прогнозом. Указанный ген был недавно идентифицирован как глицерол-3-фосфатдегидрогеназа 1-го типа (GPD1L). Было показано, что мутация в GPD1L приводит к частично-му снижению тока INa.
Данные, полученные до настоящего времени из генетических анализов, указывают на то, что эффект идентификации специфических мутаций как для постановки диагноза, так и для прогнозирования незначительный. Нет четкой информации об активных зонах (hotspots) и мутациях в гене SCN5A. До конца не выяснено, какие из найденных мутаций связаны с повышенным риском аритмических событий и ВСС. Целью генетических анализов является содействие в постановке клинического диагноза, раннем выявлении родственников с потенциальным риском ВСС, а также развитие научного прогресса в понимании гено-, фенотипической связи.
Усиление эпикардиальной и трансмуральной дисперсии реполяризации в присутствии генетических дефектов, физиологических факторов и фармакологических влияний приводит к акцентуации волны J и возможной потере ПД, содействуя тем самым возникновению экстрасистол и развитию механизма reentry во 2-й фазе ПД [13, 49].
Парасимпатические агонисты, такие как ацетилхолин, способствуют потере «свода» ПД [29] вследствие угнетения ICa и/или усиления калиевого тока. Агонисты бета-адренергических рецепторов восстанавливают «свод» ПД, так как они усиливают ICa. Блокаторы натриевых каналов так-же способствуют потере «свода» ПД ПЖ собак в связи с негативным изменением вольтажа в начале 1-й фазы. Эти наблюдения согласуются с тем фактом, что у пациентов с СБ усиливается подъем сегмента ST при проведении у них вагусных проб или введении антиаритмических препаратов I класса.
Кроме того, у этих пациентов наблюдается нормализация сегмента ST после введения бета-адренергических стимуляторов и ингибиторов фосфодиэстеразы III. Потеря «свода» ПД возникает быстрей в эпикарде ПЖ, чем ЛЖ собак вследствие более выраженной 1-й фазы (зависимой от Ito) ПД в эпикарде ПЖ. Эта разница объясняет тот факт, что СБ – это болезнь ПЖ.
Ранее предполагалось, что для симулирования СБ и получения аритмогенного субстрата необходимо снизить ток через натриевые каналы. Согласно результатам недавних экспериментальных исследований, выполненных на участке миокарда с артериальной перфузией, комбинация блокады INa и ICa является более эффективной, чем блокада лишь INa.
Для объяснения подъема сегмента ST при СБ можно выдвинуть две гипотезы: замедление проведения в эпикарде выводного отдела ПЖ [37] и/или усиление ПД эпикарда ПЖ, что может привести к потере «свода» ПД.
Сегмент ST в основном является изоэлектрическим в связи с отсутствием трансмурального градиента вольтажа на уровне плато ПД. Акцентуация зазубрины на ПД ПЖ в физиологических условиях приводит к усилению трансмурального градиента вольтажа, что вызывает появление выраженной волны J или подъема точки J на электрокардиограмме. Если реполяризация эпикарда предшествует реполяризации клеток в зонах М и эндокарда, то зубец Т сохраняется положительным. Это проявляется реполяризацией типа «седла» на ЭКГ. Более выраженная акцентуация зазубрины может сопровождаться удлинением ПД эпикарда, таким образом, направление реполяризации через стенку ПЖ и трансмуральный градиент вольтажа идут в обратном направлении, приводя к развитию сегмента ST типа «свода» и инвертированию зубца Т, которые являются типичными для СБ. Замедление активации эпикарда также может способствовать инвертированию зубца Т. Наблюдаемый в эксперименте нисходящий характер сегмента ST, который часто проявляется как зубец R', приводит к появлению признаков БПНПГ. Это возникает вследствие преждевременной реполяризации эпикарда ПЖ, а не истинного нарушения в системепроводимости.
Кроме того, попытки Т. Miyazaki и соавт. показать запоздалую активацию ПЖ у пациентов с СБ не привели к успеху. Предполагается, что аритмогенный субстрат появляется в том случае, когда значительные изменения равновесия электрических токов приводят к потере «свода» лишь в отдельных зонах эпикарда. Потеря «свода» ПД в эпикарде (но не в эндокарде) приводит к развитию выраженной трансмуральной дисперсии реполяризации и рефрактерности, ответственных за появление «уязвимого окна», на протяжении которого преждевременный импульс или экстрасистола может индуцировать аритмию по механизму повторного входа. Проведение «свода» ПД от мест, в которых он поддерживается, к зонам, в которых этот «свод» исчезает, приводит к повторному локальному возбуждению по механизму reentry во 2-й фазе ПД. Это генерирует раннюю экстрасистолу (в уязвимом периоде) и приводит к развитию механизма повторного входа, вызывающего ЖТ/ФЖ. Преждевременное сокращение, возникающее во 2-й фазе ПД, сливается с собственным зубцом Т и делает его более отрицательным. Эта картина часто наблюдается перед началом полиморфной ЖТ.
Экспериментальные исследования, проведенные на миокарде с артериальной перфузией, подтверждают данную гипотезу [36]. S. A. Aiba и соавт. использовали картографическую оптическую систему высокого разрешения, которая позволила осуществлять одновременную регистрацию транс-мембранного ПД в 256 трансмуральных зонах экс-периментального миокарда. При этом было проде-монстрировано острое возникновение градиента реполяризации между зонами, в которых исчезает «свод» ПД, и теми зонами, где он сохраняется. Это приводит к генерированию ранней экстрасистолы во 2-й фазе ПД. Данное исследование показало, что механизм reentry, появляющийся сначала в эпикарде, а затем постепенно распространяющий-ся трансмурально, является ответственным за воз-никновение полиморфных ЖТ/ФЖ.
Т. Kurita и соавт. установили монофазные электроды для регистрации ПД на поверхность эпикарда и эндокарда ВТ ПЖ у пациентов с СБ и выявили наличие выраженной зазубрины на ответе эпикарда, тем самым показано наличие этого типа нарушений и у человека. Таким образом, акцентуация зазубрины ПД эпикарда лежит в основе подъема сегмента ST. Возможная потеря «свода» ПД эпикарда ПЖ еще больше увеличивает подъем сегмента ST. Reentry во 2-й фазе ПД способствует возникно-вению экстрасистолы, индуцирующей ЖТ/ФЖ при СБ. Недавно были предоставлены экспериментальные доказательства этого механизма. Аритмии (ЖТ/ФЖ), возникшие в этих экспериментальных препаратах, напоминают во многом тахикардию типа TdP. По-видимому, эта аритмогенная активность связана со спиралевидной мигрирующей волной, которая генерирует полиморфную ЖТ.
При СУИ Q–T длительность интервала Q–T из-меняется в зависимости от вводимой дозы препарата. При CБ длительность интервала Q–Tне изменяется, а при СУИ Q–T его длительность снижается при увеличении дозы препарата. Общей чертой этих двух синдромов является усиление трансмуральной дисперсии реполяризации, которая приводит к развитию тахикардии типа TdP, когда величина дисперсии достигает порога для развития механизма reentry. Этот порог снижается при снижении длительности ПД и рефрактерности.
Различие в распространенности СБ в зависимости от пола можно связать с более выраженным Ito-индуцированным ПД в эпикарде ПЖ у мужчин. Более выраженный Ito вызывает конец 1-й фазы ПД ПЖ и реполяризацию с более низкими показателями, что способствует потере ПД и развитию 2-й фазы reentry и полиморфной ЖТ.
ТРАНСМУРАЛЬНЫЕ КЛЕТОЧНЫЕ И ИОННЫЕ ИЗМЕНЕНИЯ
Для нормального сцепления между возбуждением и сокращением, а также для поддержания сердечного ритма необходима координированная деятельность ионных каналов сердечной мышцы.
Анкирины – это внутриклеточные полипепти-ды, необходимые для биосинтеза и поддержания мембранных доменов в возбудимых и невозбудимых клетках. Они являются мембранными адаптивными молекулами, которые играют важную роль во взаимодействии интегральных мембранных белков и спектриновой сети цитоскелета. Это цитоскелетный белок, адаптирующий и контролирующий выход Ca2+ в инозитол 1, 4, 5-трифосфатных рецепторах (IP(3)R) эндоплазматического ретикулума. Семейство анкириновых («anchor» – от англ. «якорь») полипептидов участвует в распределении-прикреплении и стабилизации мембранных белков, включая ионные каналы, переносчики, ионообменники и клеточные адгезивные молекулы в различных тканях и типах клеток организма: эритроцитах, почках, легких и головном мозге.
Мутация генов анкирина приводит к такому тяжелому наследственному заболеванию, как фатальный аритмический синдром. При этом есть два анкириновых генных продукта – 220 kDa анкирин-Б, также известный как анкирин 2, и 190 kDa анкирин-Г, связанных с распределением-прикреплением различных мембранных ионных каналов и переносчиков к возбуждаемым мембранным доменам кардиомиоцитов.
Анкирин-Б и анкирин-Г – составные части сердечной мышцы, которые прикрепляют различные ионные каналы, насосы, переносчики к физиологическим сайтам воздействия. В экспериментах с мышами мутации анкирина-Б вызывают сердечный аритмический синдром, связанный с нарушением кальциевого гомеостаза.
Анкирин-Г связан с основными потенциалоза-висимыми сердечными натриевыми каналами, и потеря этого взаимодействия вследствие мутации Na(v)1.5 приводит к СБ.
С.А. Mohler и соавт. выявили человеческую генетическую мутацию (E1053K) в связанных с анкирином Na(v)1.5, которая приводит к СБ. Мутация E1053K препятствует связыванию Na(v)1.5 и анкирина-Г, а также предотвращает накопление Na(v)1.5 на клеточной поверхности желудочковых кардиомиоцитов. Анкирин-Г и Na(v)1.5 находятся во вставочных дисках и T-трубочках мембран кардиомиоцитов, Na(v)1.5 преципитирует с анкирином-Г в растворе лизата сердца крыс. Приведенные данные предполагают наличие связи между Na(v)1.5 и анкирином-Г и показывают, что анкирин-Г необходим для локализации Na(v)1.5 в возбуждаемых мембранах кардиомиоцитов. Согласно результатам исследований нейронов и кардиомиоцитов, анкирин-Г участвует в общем механизме локализации потенциалозависимых Na(v)-каналов в местах функционирования различных возбудимых клеткок. Анкирин-Г необходим для экспрессии сер-дечных потенциалозависимых Na(v)-каналов, Na(v)1.5 в специализированных доменах мембран сердечных клеток, его мутация приводит к потере распределения Na(v)1.5 на клеточной по-верхности кардиомиоцитов. Изменения в SCN5A (кодирует Na(v)1.5) у человека, которые блокируют взаимодействие Na(v)1.5 с анкирином-Г, приводят к нарушению мембранной экспрессии Na(v)1.5 и CБ [7].
СОБСТВЕННАЯ ГЕТЕРОГЕННОСТЬ ЖЕЛУДОЧКОВОГО МИОКАРДА
В миокарде желудочков имеются по крайней мере три типа клеток с различными электрофизиологическими свойствами: эпи- и эндокардиальные клетки, а также клетки М [46]. Эти три типа клеток различаются по своим реполяризационным характеристикам в фазах 1 и 3 ПД. Эпикарди-альные и М-клетки в основном имеют выраженную 1-ю фазу, которая обусловлена выходящим током (Ito), чувствительным к 4-аминопиридину (4-АП), вследствие чего потенциал действия на электрокардиограмме приобретает форму «пика», «свода» или зазубрины. Эта региональная разница Ito была выявлена в желудочковых миоцитах собак, кошек, кроликов, крыс и человека.
Между эпикардиальными и М-клетками левого и правого желудочков существует разница в величине зазубрины ПД и соответствующая разница в Ito. Это объясняет тот факт, что СБ является заболеванием именно ПЖ.
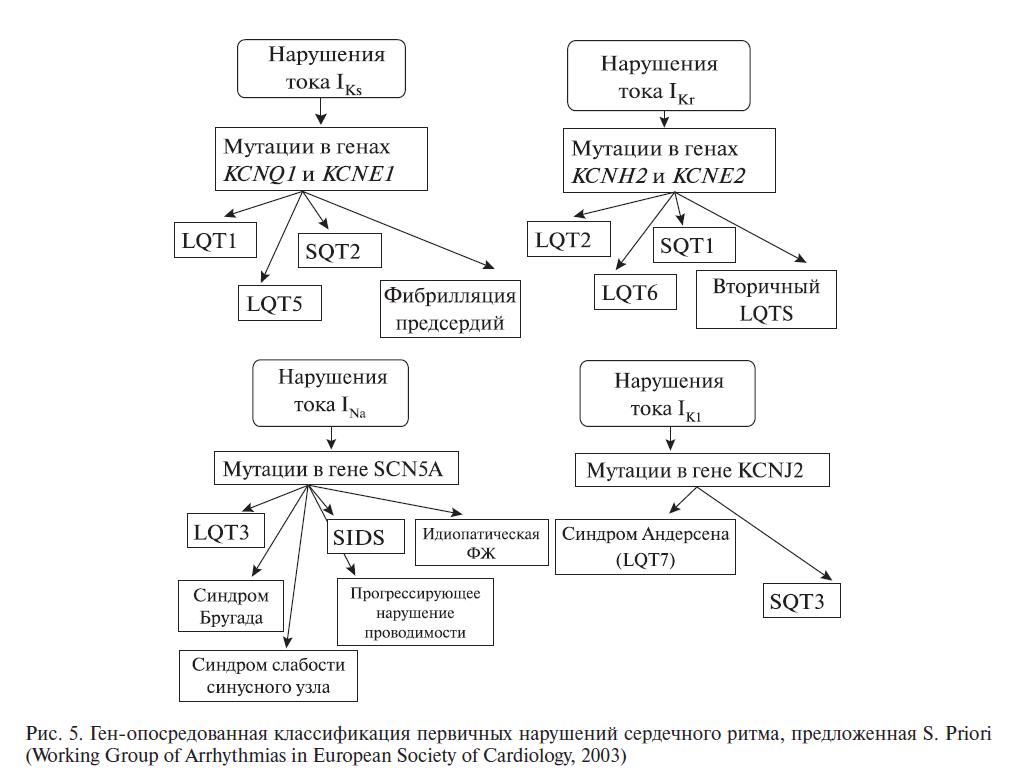
Между слоями эпи- и эндокардиальных клеток находятся переходные и М-клетки. Отличительным признаком М-клеток является то, что в ответ на замедление ЧСС и/или при введении препаратов, увеличивающих длительность ПД, их ПД увеличивается в большей степени, чем в других клетках. У собак ионной основой этих отличительных характеристик М-клеток являются: наличие позднего выпрямляющего тока медленной активации (IKs), более выраженный поздний ток натрия (INa) и более выраженный ток Na-Ca-обмена. В эксперименте с сердцем собаки было показано, что поздний выпрямляющий ток быстрой активации (IKs) и входящий выпрямляющий ток (IK1) подобны во всех трех типах трансмуральных клеток. Была описана трансмуральная и апикобазальная разница плотности каналов IKr в сердце хорьков, при этом ток IKr и белки канала более развиты в эпикарде. В сердце собаки наблюдается более выраженный ток IKs в изолированных М-клетках ПЖ, чем ЛЖ. ICa между изолированными клетками эпикарда, эндокарда и М-клетками стенки левого желудочка существенным образом не отличается. Однако в одном исследовании была найдена разница свойств каналов Ca2+ желудочковых эндо- и эпикардиальных клеток собак и показано, что ICa является более выраженным в миоцитах эндокарда, чем эпикарда. Также во всех эндокардиальных миоцитах был зарегистрирован низкопороговый ток Ca быстрой активации и дезактивации, подобный току типа Т, в то время как в эпикардиальных миоцитах этот ток не был особенно выраженным или отсутствовал. Хотя М-клетки по своим гистологическим свойствам похожи на эпи- и эндокардиальные клетки, их электрофизиологические и фармакологические свойства являются «гибридными между клетками Пуркинье и желудочков. Так же как и клетки Пуркинье, М-клетки вызывают выраженное удлинение ПД и отвечают преждевременной постдеполяризацией в ответ на блокаторы IKr, тогда как эпи- и эндокардиальные клетки не имеют этого свойства. Подобно клеткам Пуркинье, М-клетки отвечают поздними постдеполяри-зационными потенциалами в ответ на препараты, вызывающие нагрузку и перегрузку кардиомиоцитов кальцием, эпи- и эндокардиальные клетки не обладают этим свойством. В отличие от клеток Пуркинье, М-клетки удлиняют свой ПД в ответ на блокаторы IKs, эпи- и эндокардиальные клетки также удлиняют свой ПД в ответ на указанные блокаторы. Клетки Пуркинье и клетки М имеют различный ответ на адренергические альфа-агонисты. Стимуляция рецепторов альфа-1 приводит к увеличению длительности ПД в волокнах Пур-кинье, укорочению ПД в М-клетках и лишь в незначительной степени влияет на ПД эндо- и эпикардиальных клеток (рис. 5).
В стенке ЛЖ сердца собаки было детально исследовано размещение клеток М, имеющих более продолжительный ПД (BCL > 2000 мс). Они зачастую локализуются между глубокими слоями субэндокарда и средними слоями миокарда передней [45] и латеральной стенок, а также на протяжении всей стенки выводного отдела ПЖ, в то время как переходные клетки размещаются во всей стенке ЛЖ. М-клетки также присутствуют в глубоких слоях эндокарда, включая папиллярные мышцы, трабекулы и межжелудочковую перегородку. В отличие от клеток Пуркинье, М-клетки не найдены в разрозненных пучках или островках миокарда, хотя и существуют доказательства их локализации в отдельных мышечных слоях. Клетки с подобными характеристиками М-клеток были также открыты у собак, гвинейских свинок, кроликов, свиней и у человека [8].
В ряде исследований показано, что активация парасимпатической или торможение симпатической нервной системы усиливает аритмогенез. Действительно, в 93,3% случаев синкопальные приступы при СБ возникают в вечернее и ночное время (с 18 до 06 ч), причем чаще во второй половине ночи. Кроме того, в патогенезе СБ играет роль

дисбаланс между б- и в-адренергической стимуля-цией. Так, при введении пациентам б-адреноми-метиков или в-адреноблокаторов наблюдалось увеличение подъема сегмента ST, а при введении б-адреноблокаторов и в-адреномиметиков – его уменьшение [19]. Первое описание волны J было сделано в 20-х годах прошлого века, с помощью экспериментов на животных с гиперкальциемией [47]. Более де-тальное описание и характеристику этой волны представил через 30 лет Осборн, с помощью экс-периментальной гипотермии у собак. В клинической практике волна J появляется при гипотермии и гиперкальциемии. Волна J, возникающая при гипотермии, – это результат выраженного акцентирования морфологии «пика» и «свода» потенциала действия М-клеток и клеток эпикар-да (то есть увеличение ширины и высоты зазуб-рины). Кроме индуцирования более выраженной зазубрины гипотермия приводит к замедлению проводимости, отсюда – возможность раздельного появления зубца J и комплекса QRS. Акцентирование зубца J при гиперкальциемии также может быть объяснено на основании акцентирования зазубрины ПД эпикарда вследствие усиления тока хлора, активированного кальцием и снижением ICa. Выраженность зазубрины также лежит в основе электрокардиографических и аритмогенных проявлений СБ. Так как считается, что СБ является следствием чрезмерного увеличения волны J, следует обсудить электрические основы данной волны. На-личие зазубрины на ПД эпикарда и ее отсутствие на ПД эндокарда приводит к трансмуральному градиенту во время желудочковой активации, который выявляется как поздняя волна дельта после комплекса QRS волной J или зубцом Осборна. Часто можно обнаружить волну J на ЭКГ собак или бабуинов. У человека, как правило, об-наруживается подъем точки J, а не отличительная волна J. Считается, что выраженная волна J на человеческой электрокардиограмме является патогномоничным признаком гипотермии или гиперкальциемии.
Большие или анормальные волны J на протя-жении длительного времени связывались с идиопатической ФЖ, а также с СБ [39]. При СБ волна J проявляется подъемом сегмента ST в правых грудных отведениях ЭКГ. В некоторых исследованиях была подчеркнута схожесть между условиями, предрасполагающими к повторному входу во 2-й фазе, и признаками, сопутствующими СБ. Как было отмечено выше, потеря «свода» ПД в эпикарде, но не в эндокарде генерирует трансмуральный ток, проявляющийся на ЭКГ элевацией сегмента ST, подобной таковой при СБ. Недавно B. D. Thomsen и соавт. подтвердили возникновение повторного входа во 2-й фазе ПД у человека.
На изолированных миоцитах эпикарда стенки ЛЖ кролика была показана бóльшая плотность тока хлора, активированного ц-АМФ, чем тока хлора в миоцитах эндокарда. В настоящее время считается, что Ito2, который раньше объяснялся калиевым током, на самом деле является током хлора, активированного кальцием, который также влияет на зазубрину ПД, но пока не известно, различаются ли эти токи между тремя типами клеток желудочкового миокарда.
Трансмуральная активация внутри тонкой стенки ПЖ является относительно быстрым процессом. Это приводит к тому, что волна J «покрывается» комплексом QRS. И хотя зазубрина на ПД более выраженная в эпикарде ПЖ, этот желудочек лишь в незначительной степени влияет на появление волны J в нормальных условиях. Эти наблюдения согласуются с появлением волны J в тех отведениях ЭКГ, в которых суммарный электрический вектор направлен трансмурально через ЛЖ и перегородку. Вследствие этого волна J у собак более выражена в отведениях II, III, aVR, aVF, а также в средних и левых грудных отведениях V3–V6. Подобная картина наблюдается и на человеческой электрокардиограмме. Кроме того, с помощью векторокардиограммы было показано, что волна J образует дополнительную петлю, возникающую на соединении петли QRS и T. Она направлена влево и вперед, что объясняет выраженность данного зубца в отведениях, соответствующих ЛЖ.
Пространственная дисперсия реполяризации, выражающаяся в трансмуральной, транссептальной и апикобазальной ее дисперсии, генерирует градиенты вольтажа, способные вызывать формирование волны J и зубца Т на ЭКГ. Усиление этой дисперсии лежит в основе жизнеугрожающих желудочковых аритмий, связанных с наследственными ионными «каналопатиями», включающими СУИ Q–T, СКИ Q–T, СБ, а также полиморфную желудочковую катехоламинергическую тахикардию (ПЖКТ). При СУИ Q–T усиление трансмуральной дисперсии реполяризации (ТДР) зачастую является следствием удлинения ПД М-клеток, в то время как при СБ возникает селективное укорочение ПД эпикарда ПЖ. По-видимому, укорочение ПД эндо- или эпикарда является ответственным за усиление ТДР СКИ Q–T. В случае ПЖКТ причи-ной усиления ТДР является обратное направление активации желудочковой стенки.
Таким образом, СУИ Q–T, СКИ Q–T, СБ, ПЖКТ – это патологии с различными фенотипами и этиологическими факторами, но с общим механизмом возникновения ВСС.
АЛЬТЕРНАЦИЯ ВОЛНЫ Т
Одним из менее известных, но наиболее информативных предикторов высокого риска ВСС является определение альтернации зубца Т при проведении пробы с физической нагрузкой или по данным суточного мониторирования ЭКГ [34]. Альтернация зубца Т – это числовая характеристи-ка изменений морфологии зубца Т, проявляющихся на электрокардиограмме в виде чередования комплексов QRS различных типов. При проведении тестов с физической нагрузкой альтернация зубца Т представляется как снижение амплитуды или полярности зубцов Т и изменение морфологии сегмента ST в разных комплексах. Первые исследования диагностической значимости альтернации зубца Т были проведены D. Rosenbaum и соавт. Авторы выявили сильную зависимость между величиной альтернации зубца Т и частотой развития желудочковых аритмий по данным электро-физиологического исследования (ЭФИ).
По результатам работы был сделал вывод, что альтернация зубца Т служит одним из маркеров возникновения ВСС. Исследования последних лет доказа-ли высокую информативность метода альтернации зубца Т для определения группы пациентов, нуждающихся в имплантируемом кардиовертере-дефибрилляторе (ИКВД). В работе D. Predescu и соавт. установлена высокая чувствительность метода альтернации зубца Т по сравнению с ЭФИ для оп-ределения риска возникновения желудочковых аритмий. До 10% всех случаев ВСС обусловлены наследственной патологией, ведущей к возникно-вению опасных для жизни аритмий. Наиболее распространены СУИ Q–T и СБ. Оба заболевания являются частыми причинами летальности в детском возрасте. Для оценки риска ВСС у детей с наследственной патологией наиболее простым и достоверным методом исследования является альтернация зубца Т при проведении теста с физической нагрузкой. До недавнего времени единственным методом оценки альтернации зубца Т был спектральный метод, основанный на анализе вариаций морфологии зубца Т в частотной области. Новая программа, разработанная компанией «GE Healthcare IT», предназначена для измерения альтернации зубца Т непосредственно во временной области, что не требует поддержания высоких значений ЧСС в течение продолжительного времени. Это особенно важно у пациентов с низкой фракцией выброса ЛЖ. Таким образом, метод оп-ределения альтернации зубца Т помогает прогнозировать риск возникновения опасных для жизни желудочковых аритмий и своевременно выбрать правильную тактику лечения пациентов с любой патологией сердечно-сосудистой системы. Исследования показали, что во время альтернации Т-волны запускаются два механизма – потеря ПД эпикарда и скрытая 2-я фаза reentry [16].
ПРИОБРЕТЕННЫЙ СИНДРОМ БРУГАДА
В литературе встречаются указания на появление типичных для СБ изменений на ЭКГ под воздействием других причин, помимо генетической патологии. За 15 лет изучения СБ накопились данные о приобретенном СБ. Описаны бругадоподобные изменения ЭКГ у больного во время лихорадки, которые, однако, не были подтверждены после нормализации температуры при выполнении теста с введением антиаритмических препаратов I класса. Изменения на ЭКГ по типу бругадоподобных, без клинического подтверждения синдрома, могут наблюдаться у пациентов с экскавацией грудной клетки (вероятно, из-за хронического поврежде-ния ПЖ), при гемоперикарде.
Описан случай регистрации типичной для СБ ЭКГ-картины при опухоли средостения с нормализацией ЭКГ после ее удаления, что, возможно, связано со сдавлением опухолью ВТПЖ.
Имеются сообщения о появлении признаков этого синдрома при употреблении больших доз кокаина, трициклических антиде-прессантов [4], при гиперкалиемии, гиперкальциемии, дефиците тиамина, гиперпаратиреоидизме, гипертестостеронемии, аритмогенной дисплазии ПЖ, перикардите, инфаркте миокарда, стенокардии Принцметала, механической обструкции ВТ ПЖ, тромбоэмболии легочной артерии, расслаивающейся аневризме аорты, различных ано-малиях центральной и вегетативной нервной системы, мышечной дистрофии Дюшенна, атаксии Фредерика. Однако трудно судить, являются ли они проявлением скрытого СБ или приобретенными бругадоподобными изменениями.
Прием лекарственных препаратов, ингибирующих натриевые каналы, также может вызывать СБ. Лекарственно индуцированный СБ описан при лечении месалазином, ваготоническими препаратами, бета-адренергическими агонистами, бета-адреноблокаторами, антигистаминными препаратами 1-го поколения, антималярийными средствами, седативными, антиконвульсантами, нейролептиками, три- и тетрациклическими антидепрессантами, препаратами лития.
По данным экспериментов in vitro и in vivo, сек-ционных и клинических исследований антиконвульсанты, антидепрессанты, нейролептики, анксиолитики и нормотимики способны блокировать быстрые калиевые HERG-каналы, натриевые каналы (вследствие дефекта гена SCN5A) и кальциевые каналы L-типа, вызывая таким образом функциональную недостаточность всех каналов сердца [42]. Имеющиеся в медицинской литературе сообщения о СБ, тесно ассоциированном с приемом психотропных препаратов, касаются в основном суицидов, случаев передозировки и интоксикации, то есть токсических доз лекарственных средств [10].
Ускоренная инактивация натриевых каналов, вызванная мутациями гена SCN5A (связанными с СБ), как показали исследования, сопровождалась повышением температуры. Действительно, несколько случаев демонстрируют, что лихорадка может способствовать выявлению латентной формы СБ и вызвать ЖТ. P. C. Keller и соавт. выявили мутацию F1344S в гене SCN5A у пациентов с СБ и ЖТ, вызванной лихорадкой.
В повседневной практической работе мы наблюдаем появление БПНПГ необычной формы (с умеренным подъемом сегмента ST) – «saddle-back type» у пациентов, принимающих психотропные препараты в дозах терапевтического стандарта [23]. При этом регистрация таких бругадоподобных изменений на ЭКГ сопровождает ухудшение психосоматического состояния больных по типу «чрезмерной транквилизации», с симптомами общей слабости, сонливости, гипотонии, брадикардии, что напоминает клиническую картину глубокой гиперседации. После коррекции терапии или отмены психотропного препарата наблюдается быстрая обратная динамика ЭКГ-изменений вслед за улучшением клинического состояния больных.
ЛЕЧЕНИЕ ПАЦИЕНТОВ С СИНДРОМОМ БРУГАДА
Лечение СБ проводят с учетом клинических особенностей заболевания, характера генетических нарушений (наличие дефектного белка Na-каналов). При наличии клинических симптомов больным необходима имплантация кардиоверте-ра-дефибриллятора (КВД), такой же метод лечения нужен больным с индуцированной при ЭФИ ЖТ.
Из антиаритмических препаратов необходимо назначать препараты IA класса (хинидин и дизопирамид) или амиодарон. Следует избегать назначения препаратов I класса, таких как новокаинамид, флекаинид, аймалин, которые, изолированно блокируя натриевый ток, провоцируют манифестацию СБ. Препараты класса IС, такие как флекаинид и пропафенон, и класса IА, такой как прокаинамид, противопоказаны, потому что они способствуют выявлению скрытого СБ и вызывают аритмогенез.
До настоящего времени медикаментозная терапия при СБ не нашла широкого применения в связи с отсутствием препаратов, достоверно снижающих смертность таких больных. Имеются единичные указания на эффективность предупреждения желудочковых нарушений ритма приназначении пропранолола и дизопирамида, хотя они могут приводить и к еще более выраженному подъему сегмента SТ. Дизопирамид (класс IA) в некоторых случаях нормализует элевацию сегмента ST, а в некоторых также выявляет скрытый СБ [48].
Есть наблюдение, свидетельствующее о предотвращении рецидивирования ФЖ при внутривенном введении изопротеренола. Предпринимались попытки назначения пациентам с СБ амиодарона, однако, по мнению братьев Бругада, прием амиодарона и b-блокаторов не предупреждает развитие ВСС у таких больных (26% умерли в течение 3 лет, что достоверно не отличается от смертности больных, не получавших лечение, – 31%).
В настоящее время ведется поиск других препаратов, которые могли бы быть использованы для лечения этого синдрома. Например, описан случай предотвращения регулярных эпизодов ФЖ при оральном применении цилостазола (ингибитора фосфодиэстеразы), что было подтверждено при проведении теста периодической отмены. Уменьшают подъем сегмента ST (без указания эффективности предот-вращения аритмий) катехоламины, b-адреномиметики и a-адреноблокаторы.
Наличие выраженного Itо является составляющей механизма развития СБ. Следовательно, самый лучший подход заключается в ингибировании Itо. Кардиоселективные и Ito-специфичные блокаторы в настоящее время не доступны. 4-аминопиридин в низких концентрациях является специфичным для этих каналов, подавляет аритмогенез при пограничных видах СБ, но он не является кардиоселективным, блокирует Itо также в нервной системе и не улучшает клиническую картину из-за его неблагоприятного влияния на нервную систему [21].
Известно, что хинидин в низкой дозе (300–600 мг) может предотвратить электрофизиологическую индукцию ЖТ и является дополнительной терапией у пациентов с разряженным КВД [15].
Поиски кардиоселективного и Itо-специфичного блокатора продолжаются. Еще один препарат, рассматриваемый для этих целей, – тедисамил, который в настоящее время используется для лечения ФП. Тедисамил может оказаться более эффективным, чем хинидин. Тедисамил и хинидин способны подавлять механизм развития СБ путемблокирования Itо. Но в то же время они блокируютспецифические каналы, что способствует возникновению СУИ Q–T. Таким образом, эти агенты могут заменить возникновение одной формы полиморфной ЖТ другой, особенно при брадикардии и гипокалиемии (способствуют возникновению TdP). Однако большинство пациентов с СБ – это здоровые мужчины, для которых риск возникновения препарат-индуцированной TdP низкий.
При использовании высоких доз хинидина(1000–1500 мг/день) происходит блокирование Itо, но TdP не развивается.
Еще один препарат, который является блокатором Itо и I-каналов, – это AVE0118 [40]. Его преимущество в том, что он не блокирует I-каналы, не удлиняет интервал Q–T и не индуцирует развитие TdP. Недостаток этого препарата состоит в том, что он метаболизируется в печени и не эффективен при оральном приеме.
Также эффективен бета-адренергический пре-парат изопротеренол, который увеличивает поток кальция [27]. В некоторых случаях использование изопротеренола в комбинации с хинидином нормализует элевацию сегмента ST, особенно у детей. Новым препаратом является фосфодиэстераза (ингибитор килостазола), которая нормализует сегмент ST путем увеличения кальциевого потока и уменьшения Itо.
Единственным достоверно эффективным методом лечения больных с симптомным вариантом СБ в настоящее время считается ИКВД, предотвращающий эпизоды ВСС. По некоторым дан-ным, прием амиодарона при наличии имплантированного кардиовертера-дефибриллятора может уменьшить частоту его разрядов (хотя эффективность амиодарона в предотвращении аритмий, как было отмечено выше, не доказана).
Вопрос об имплантации КВД пациентам с бессимптомным вариантом СБ в настоящее время всесторонне обсуждается.
Есть данные, что предиктором перехода бессимптомного СБ в симптомный может служить также более длинный интервал Н–V(59±8 мс против 48±11 мс) [26]. Помимо названных показаниями к имплантации КВД у бессимптомных пациентов в настоящее время считаются: мужской пол, возраст 30–40 лет, семейный анамнез по ВСС, наличие мутации в гене SCN5A, наличие спонтанных изменений в отве-дениях V1– V3 на ЭКГ.
Для предупреждения ФЖ при СБ используются классические антиаритмические препараты, обес-печивающие эффект в 60% случаев [5]. Генетичес-ки детерминированное поражение натриевых каналов теоретически предполагает меньшую эффективность препаратов 1-й группы, а также возможность проявления проаритмогенного эффекта при их использовании. Согласно алгоритму формирования антиаритмической терапии, известному под названием «сицилианский гамбит», антиаритмическими препаратами, обеспечиваю-щими активную блокаду натриевых каналов, являются новокаинамид, дизопирамид, хинидин, ритмонорм, гилуритмал, флекаинид, энкаинид. Менее выраженное блокирующее действие отмечено у лидокаина, мексилетина, токаинида, бепридила, верапамила, кордарона и обзидана.
Можно предположить, что при СБ более безопасно применять препараты, не блокирующие натриевые каналы, – дилтиазем, бретилиум, соталекс, надолол (коргард). Однако целенаправленных ис-следований в этой области пока не проводилось.
Медикаментозные подходы к лечению СБ активно разрабатываются, но результаты этих исследований остаются противоречивыми. Убедительных согласованных данных за эффективность каких-либо лекарственных препаратов в длительном предотвращении приступов ЖТ/ФЖ в настоящее время практически нет. Для пациентов с высоким риском ВСС методом лечения, увеличивающим продолжительность жизни, является имплантация КВД.
ЛИТЕРАТУРА
1. Бокерия, Л. А. Синдром Бругада – клинико-диагностичес-кие критерии и лечение / Л. А. Бокерия, А. Ш. Ревишвили, И. В. Проничева // Анналы аритмол. – 2005. – № 4. – С. 35.
2. Иванов, Г. Г. Основные механизмы, принципы прогноза и профилактики внезапной сердечной смерти / Г. Г. Иванов, А. С. Сметнев, А. Л. Сыркин и др. // Кардиология. – 1998. – № 12. – С. 64–73.
3. Макаров, Л. М. Синдром Бругада / Л. М. Макаров // Ме-дицина неотложных состояний. – 2008. – № 1 (14). – С. 96–99.
4. Фомина, И. Г. Значение альтернации зубца Т для диагнос-тики опасных для жизни аритмий / И. Г. Фомина, А. И. Тарзиманова // Кардиология. – 2006. – № 6. – С. 90–91.
5. Alings, M. «Brugada» syndrome: Clinical data and suggested pathophysiological mechanism / M. Alings, A. Wilde // Circulation. – 1999. – Vol. 99. – P. 666–673.
6. Antzelevitch, C. Brugada syndrome. Report of the second con-sensus conference / C. Antzelevitch, P. Brugada, M. Borggrefe et al. // Circulation. – 2005. – Vol. 111. – P. 659–670.
7. Antzelevitch, C. Cellular mechanisms underlying the Brugada syndrome / C. Antzelevitch, J. Fish, J. M. Di Diego // The Brugada Syndrome: From Bench to Bedside; eds C. Antzelevitch, P. Brugada, J. Brugada, R. Brugada. – Oxford: Blackwell Futura, 2004. – P. 52–77.
8. Antzelevitch, C. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells / C. Antzelevitch, S. Sicouri, S. H. Litovsky et al. // Circ. Res. – 1991. – Vol. 69. – P. 1427–1449.
9. Aramaki, K. Chest pain and ST elevation associated with fever in patients with asymptomatic Brugada syndrome Fever and chest pain in Brugada syndrome / K. Aramaki, H. Okumura, M. Shimizu // Int. J. Cardiol. – 2005. – Vol. 103. – P. 338–339.
10. Babaliaros, V. C. Tricyclic antidepressants and the Brugada syn-drome: an example of Brugada waves appearing after the admin-istration of desipramine / V. C. Babaliaros, J. W. Hurst // Clin. Cardiol. – 2002. – Vol. 25. – P. 395–398.
11. Baroudi, G. Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome / G. Baroudi, S. Acharfi, Ch. Larouche, M. Chahine // Circ. Res. –2002. – Vol. 90. – P. 11.
12. Baroudi, G. Novel mechanism for Brugada syndrome: defec-tive surface localization of an SCN5A mutant (R1432G) / G. Baroudi, V. Pouliot, I. Denjoy et al. // Circ. Res. – 2001. – Vol. 88. – P. E78–E83.
13. Bezzina, C. R. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction / C. R. Bezzina, W. Shimizu, P. Yang et al. // Circulation. – 2006. – Vol. 113. – P. 338–344.
14. Brugada, J. Brugada syndrome / J. Brugada, R. Brugada, P. Brugada // Arch. Mal. Coeur. Vaiss. – 1999. – Vol. 92. – P. 847–850.
15. Brugada, J. Right bundlebranch block and ST-segment eleva-tion in leads V1 through V3. A marker for sudden death in
patients without demonstrable structural heart disease / J. Brugada, R. Brugada, P. Brugada // Circulation. – 1998. – Vol. 7. – P. 457–460.
16. Brugada, P. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and elec-trocardiographic syndrome: a multicenter report / P. Brugada, J. Brugada // J. Am. Coll. Cardiol. –1992. – Vol. 20. – P. 1391–1396.
17. Brugada P. Variability of the diagnostic ECG pattern in an ICD patient population with Brugada syndrome / P. Brugada, A. Sarkozy // J. Cardiovasc. Electrophysiol. – 2009. – Vol. 20. – P. 69–75.
18. Brugada, R. Use of intravenous antiarrhythmics to identify con-cealed Brugada syndrome / R. Brugada // Curr. Control. Trials. Cardiovasc. Med. – 2000. – Vol. 11. – P. 45–47.
19. Burashnikov, A. Differences in the electrophysiologic response of four canine ventricular cell types to a1-adrenergic agonists / A. Burashnikov, C. Antzelevitch // Cardiovasc. Res. – 1999. – Vol. 43. – P. 901–908.
20. Chen, S. M. Brugada syndrome without mutation of the cardiac sodium channel gene in a Taiwanese patient / S. M. Chen, С. Т. Kio, K. H. Lin, F. T. Chiang // J. Formos. Med. Assoc. – 2001. – Vol. 99. – P. 860–862.
21. Chinushi, M. Discrepant drug action of diso-pyramide on ECG abnormalities and induction of ventricular arrhythmias in a patient with Brugada syndrome / M. Chinushi, Y. Aizawa, Y.Ogawa et al. // J. Electrocardiol. – 1997. – April. – P. 133–136.
22. Clinicay Maternidad Suizo, Воротняк А. Д. Синдром Бруга-да // Medicus Amicus. – 2007. – № 3. – C. 45.
23. Crumb, W. J. Cardiac ion channel blocking profile of olanzap-ine and other antipsychotics / W. J. Crumb, C. Beasley, A. Thornton et al. // 38th American College of Neuro-psychopharmacology Annual Meeting. – Acapulco, 1999.
24. Di Diego, J. M. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males / J. M. Di Diego, J. M. Cordeiro, R. J. Goodrow et al. // Circulation. – 2002. – Vol. 106. – P. 2004–2011.
25. Dumaine, R. Ionic mechanisms responsible for the electrocar-diographic phenotype of the Brugada syndrome are temperature dependent / R. Dumaine, J. A. Towbin, P. Brugada et al. // Circ. Res. – 1999. – Vol. 85. – P. 803–809.
26. Fish, J. M. AVE0118, an and blocker, suppresses VT/VF in an experimental model of the Brugada syndrome / J. M. Fish, F. Extramiana, C. Antzelevitch // Circulation. – 2004. – Vol. 110, № 17. – P. 1II–193 (Abstract).
27. Fish, J. M. Tedisamil abolishes the arrhythmogenic substrate responsible for VT/VF in an experimental model of the Brugada syndrome / J. M. Fish, F. Extramiana, C. Antzelevitch // Heart Rhythm. – 2004. – Vol. 1 (1S). – P. 58 (Abstract).
28. Furuhashi, M. Prevalence of asymptomatic ST segment eleva-tion in right precordial leads with right bundle branch block (Brugada-type ST shift) among the general Japanese population / M. Furuhashi, K. Uno, K. Tsuchihashi et al. // Heart. – 2001. – Vol. 86. – P. 161–166.
29. Furukawa, Y. K+-channel blocking and anti-muscarinic effects of a novel piperazine derivative, INO 2628, on the isolated dog atrium / Y. Furukawa, K. Akahane, Y. Ogiwara, S. Chiba // Eur. J. Pharm. – 1991. – Vol. 193. – P. 217–222.
30. Gima, K. Ionic current basis of electrocardiographic waveforms: a model study / K. Gima, Y. Rudy // Circ. Res. – 2002. – Vol. 90. – P. 889–896.
31. Hong, K. Cryptic 5' splice site activation in SCN5A associat-ed with Brugada syndrome / K. Hong, A. Guerchicoff, G. D. Pollevick et al. // J. Mol. Cell. Cardiol. – 2005. – Vol. 38. – P. 555–560.
32. Kakishita, M. Mode of onset of ventricular fibrillation in patients with Brugada syndrome detected by implantable cardioverter defibrillator therapy / M. Kakishita, Т. Kurita, K. Matsuo et al. // J. Am. Coll. Cardiol. – 2000. – Vol. 36. – P. 1646–1653.
33. Kasanuki, H. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease /
H. Kasanuki, S. Ohnishi, N. Matuda, T. Nirei // Circulation. – 1997. – Vol. 95. – P. 2277–2285.
34. Kraus, F. Ueber die wirkung des kalziums auf den kreislauf / F.Kraus // Dtsch. Med. Wochenschr. – 1920. – Bd. 46. – S. 201–203.
35. Kurita, Т. The electrophysiologic mechanism of ST-segment ele-vation in Brugada syndrome / Т. Kurita, W. Shimizu, M. Inagaki et al. // J. Am. Coll. Cardiol. – 2002. – Vol. 40. – P. 330–334.
36. Litovsky, S. H. Differences in the electrophysiological response of canine ventricular subendocardium and subepicardium to acetylcholine and isoproterenol. A direct effect of acetylcholine in ventricular myocardium / S. H. Litovsky, C. Antzelevitch // Circ. Res. – 1990. – Vol. 67. – P. 615–627.
37. Lukas, A. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia / A. Lukas, C. Antzelevitch // Cardiovasc. Res. – 1996. – Vol. 32. – P. 593–603.
38. Matsuo, K. Mode of onset of ventricular fibrillation in patients with Brugada syndrome detected by implantable cardioverter defibrillator therapy / K. Matsuo et al. // J. Am. Coll. Cardiol. – 2000. – Vol. 36. – P. 1646–1653.
39. Miyazaki, T. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome / T. Miyazaki, H. Mitamura, S. Miyoshi et al. // J. Am. Coll. Cardiol. – 1996. – Vol. 27. – P. 1061–1070.
40. Mizusawa, Y. Effects of low-dose quinidine on ventricular tach-yarrhythmias in patients with Brugada syndrome: Low-dose quinidine therapy as an adjunctive treatment / Y. Mizusawa, H. Sakurada, M. Nishizaki, M. Hiraoka // Cardiovasc. Pharmacol. – 2006. – Vol. 47. – P. 359–364.
41. Monroe, M. H. Two-year case collection of the Brugada syn-drome electrocardiogram pattern at a large teaching hospital / M. H. Monroe, L. Littmann // Clin. Cardiol. – 2000. – Vol. 23. – P. 849–851.
42. Morita, H. T wave alternans in an in vitro canine tissue mode-lof Brugada syndrome / H. Morita, D. P. Zipes, J. Lopshire et al. // Am. J. Physiol. Heart Circ. Physiol. – 2006. – Vol. 291. – P. H421–H428.
43. Priori, S. G. Task Force on sudden cardiac death of the European Society of Cardiology / S. G. Priori, E. Allot, C. Blomstrom-Lundqvist et al. // Eur. Heart J. – 2001. – Vol. 22. – P. 1374–1450.
44. Probst, V. Monomorphic ventricular tachycardia due to Brugada syndrome successfully treated by hydroquinidine ther-apy in a 3-year-old child / V. Probst, S. Evain, V. Goumay et al. // J. Cardiovasc. Electrophysiol. – 2006. – Vol. 17. – P. 97–100.
45. Riera, A. R. P. Синдром анкирина-Б и синдром Бругада (Анкирина-Г) – два типа аритмогенных фатальных заболе-ваний / A. R. P. Riera, E. Schapachnik, S. Dubner // Medicus Amicus. – 2008. – № 1. – С. 24.
46. Shimizu, W. Mechanisms of disease: current understanding and future challenges in Brugada syndrome / W. Shimizu, T. Aiba, S. Kamakura // Nat. Clin. Pract. Cardiovasc. Med. – 2005. – Vol. 2. – P. 408–414.
47. Sicouri, S. Electrophysiologic characteristics of M cells in the canine left ventricular free wall / S. Sicouri, C. Antzelevitch // J. Cardiovasc. Electrophysiol. – 1995. – Vol. 6. – P. 591–603.
48. Tada, H. Brugada syndrome mimicked by tricyclic antidepres-sant overdose / H. Tada, C. Sticherling, H. Oral, F. Morady // J. Cardiovasc. Electrophysiol. – 2001. – Vol. 12. – P. 275 (Casa report).
49. Weiss, R. Clinical and molecular heterogeneity in the Brugada syndrome. A novel gene locus on chromosome 3 / R. Weiss, M. M. Barmada, T. Nguyen et al. // Circulation. – 2002. – Vol. 105. – P. 707–713.
50. Wichter, Т. Cardiac autonomic dysfunction in Brugada syn-drome / Т. Wichter, P. Matheja, L. Eckardt et al. // Circulation. – 2002. – Vol. 105. – P. 702–706.
51. Yan, G.-X. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation / G.-X. Yan, C. Antzelevitch // Circulation. – 1999. – Vol. 100. – P. 1660–1666.
В 1 больнице Минска врачи впервые провели операции по профилактике образования тромбов
В 1–й минской клинической больнице врачи впервые провели серию вмешательств по профилактике образования тромбов, которые опасны для жизни. Оторвавшись и попав в кровеносное русло, они могут закупорить сосуд, в том числе головного мозга. Особенность в том, что на этот раз не пришлось задействовать хирургов и применять общий наркоз. А пять пациентов, мужчины и женщины средних лет, быстрее пошли на поправку. Больничные палаты они покинули буквально на второй–третий день.
Речь о новой эндоваскулярной, внутрисосудистой методике — окклюзии ушка левого предсердия (в этом месте чаще всего и образуется тромб). Она становится буквально спасением при нарушениях ритма сердца — трепетании предсердий и мерцательной аритмии. Изначально таким пациентам врачи назначают лекарства, разжижающие кровь. «Однако у определенного процента людей эффект от их применения низкий или недостаточный. Для некоторых эти средства и вовсе небезопасны, так как могут спровоцировать не только аллергические реакции, но и кровотечения, — рассказывает заведующий ангиографическим кабинетом 1–й клиники, главный внештатный рентгенэндоваскулярный хирург комитета по здравоохранению Мингорисполкома Павел Черноглаз. — Потому выход был один — операция. Но если раньше, работая на опережение, хирурги проводили либо сложное вмешательство со вскрытием грудной клетки, либо оперировали без скальпеля при помощи эндоскопа, то сегодня появилась альтернатива».
Смысл в том, что в верхней части бедра в области паха врач делает небольшой прокол, вводит в него медицинские инструменты и под контролем рентгена ведет их через бедренную, а после полую вену до самого левого предсердия. Сюда и доставляют так называемый зонтик, или окклюдер, который раскрывается и заполняет изнутри собой ушко. Готово — надежную «заглушку» поставили! Теперь у тромба нет шанса навредить. И на все про все — только 40 — 50 минут. Кстати, расходы по такому ювелирному вмешательству полностью берет на себя государство, хотя стоимость только спасительного зонтика — около 10 тысяч у.е.
Раньше столь высокотехнологичные вмешательства были исключительно прерогативой РНПЦ «Кардиология» и Республиканского клинического медицинского центра Управления делами Президента. Теперь по–новому спасать пациентов будут и городские больницы. В 1–й минской клинике прорывной технологией владеют уже четыре специалиста.
Справка «СБ»
Мерцательной аритмией, приводящей к инсульту, страдают около 200 тысяч жителей Беларуси.
Советская Белоруссия № 93 (24723). Среда, 20 мая 2015
Автор публикации: Алла МАРТИНКЕВИЧ
